
Safa A. Sherfi
Molecular Biology Research Unit (MBRU), The National Ribat University, P.O. Box 32, Khartoum, Sudan
Hamid A. Dirar
Department of Botany and Agricultural Biotechnology, Faculty of Agriculture,
University of Khartoum, Sudan
Badr E. Hago
Molecular Biology Research Unit (MBRU), The National Ribat University, P.O. Box 32, Khartoum, Sudan
Mohamed E. Ahmed
Molecular Biology Research Unit (MBRU), The National Ribat University, P.O. Box 32, Khartoum, Sudan
Hassan A. Musa
Molecular Biology Research Unit (MBRU), The National Ribat University, P.O. Box 32, Khartoum, Sudan
Hassan Abu Aisha
Molecular Biology Research Unit (MBRU), The National Ribat University, P.O. Box 32, Khartoum, Sudan
Imadeldin E. Aradaib
Molecular Biology Research Unit (MBRU), The National Ribat University, P.O. Box 32, Khartoum, Sudan
Research Journal of Microbiology
Year: 2007 | Volume: 2 | Issue: 2 | Page No.: 163-169







ABSTRACT
The potential of the Polymerase Chain Reaction (PCR), as a means of detecting Escherichia coli (E. coli) DNA in suspected environmental samples, was studied. Using a pair of outer primers P1 and P2, selected from uidA gene, which encodes E. coli glucuronidase, the PCR-based assay resulted in amplification of a 486 base pair (bp) PCR product. E. coli strains from different environmental sources including recycled and drinking water as well as stagnant water were detected by this nested PCR-based assay. Amplification products were visualized on ethidium bromide-stained agarose gel. The sensitivity of the PCR assay was 100 fg of bacterial DNA with ethidium bromide-stained agarose gels. Using a pair of internal (nested) primers P3 and P4, the nested PCR produced a 186 bp PCR product. The nested PCR increased the sensitivity of the PCR assay by 1,000 times and specific PCR products were detected from 0.1 fg of bacterial DNA. Amplification product was not detected when the nested PCR-based assay was applied to DNA from other related bacteria including, Salmonella, Pseudomonas and Proteus or nucleic acid-free water. Application of this nested PCR-based assay to environmental samples resulted in direct detection of E. coli DNA from sewage water, tap water, drinking water at Shambat Campus, University of Khartoum, Sudan. This nested PCR-based assay should provide a rapid, sensitive and specific assay for direct detection and quantification of E. coli in environmental samples suspected to contain the organism.
PDF Abstract XML References
How to cite this article
Safa A. Sherfi, Hamid A. Dirar, Badr E. Hago, Mohamed E. Ahmed, Hassan A. Musa, Hassan Abu Aisha and Imadeldin E. Aradaib, 2007. Evaluation of Polymerase Chain Reaction for Direct Detection of Escherichia coli Strains in Environmental Samples. Research Journal of Microbiology, 2: 163-169.
URL: https://scialert.net/abstract/?doi=jm.2007.163.169
URL: https://scialert.net/abstract/?doi=jm.2007.163.169
INTRODUCTION
Diagnostic methods currently applied for detection of E. coli include conventional cultivation and subsequent identification by biochemical tests (Aradaib et al., 2005; Brenner et al., 1993; Byamukama et al., 2000; Carson et al., 2003; Chang et al., 1989). Serology is also useful for serotyping of specific strains of bacteria. The conventional bacterial isolation is time consuming, labor intensive and expensive (Chang et al., 1989; Byamukama et al., 2000). To address these problems, specific DNA probes derived from different genes of E. coli have been developed (Colle et al., 1996; Garner et al., 1988). However, the minimum amount of bacterial DNA that these DNA probes could detect was found to be in the nanogram range as determined by hybridization techniques (Colle et al., 1996; Fricker and Fricker, 1996). This low sensitivity of the recombinant DNA probes limits their application for direct detection of bacterial nucleic acid sequence present at very low amounts in environmental samples. Recently, application of Polymerase Chain Reaction (PCR) amplification technology has resulted in highly sensitive detection of different bacterial organisms (Aradaib et al., 2005; Fiksdal et al., 1994; Heininger et al., 1999). The development of a rapid, sensitive and specific PCR assay for the detection of E. coli would greatly facilitate detection of the organism in contaminated environments. In the present study, we evaluated a reproducible, sensitive and specific assay for detection of E. coli in purified colonies or directly in suspected environmental samples using nested (nPCR) amplification technology.
MATERIALS AND METHODS
Collection of Samples
Environmental samples suspected to contain E. coli were collected in sterile containers from different sources including sewage water, recycled water and drinking water. All the environmental samples were collected from different water sources at Shambat Campus of the University of Khartoum, Sudan.
Conventional Isolation and Identification
Environmental samples were grown on MacConkey’s media as selective and differential media to exclude non-lactose fermentors of the family Enterobacteriaceae. Lactose is included as a fermentable carbohydrate with a pH indicator, usually neutral red. Strong acid producers like Escherichia, Klebsiella and Enterobacter produced red colonies. Loopful of suspension from each medium was streaked on Eosin Methylene Blue (EMB) agar, plates were incubated 18-24 h at 37°C and examined for typical E. coli colonies, dark centered with or without metallic sheen. Two typical colonies from each (EMB) plate were picked and transferred to nutrient agar slants for morphological and biochemical tests. Slants were incubated at 37°C for 18-24 h. Gram stain on each culture was performed, for all cultures appearing as Gram- negative, short rods or cocci were identified by Triple Sugar Iron (TSI) biochemical scheme, motility test, indole test, urease test, lysine decomposition test and DNA hydrolysis test (Colle et al., 1996). The rest of the samples were isolated using multiple tube technique and identified as described previously (Geldreich, 1975).
Extraction of Bacterial Nucleic Acids from Environmental Samples
Extraction of bacterial nucleic acids from environmental samples was made possible using a commercially available QIAamp tissue kit (QIAGEN Inc. Hamburg, Germany) according to the manufacturer’s instructions. Briefly, (200 μL of sample, 30 μL of proteinase K stock solution and 200 μL of lysing buffer were pipetted into 1.5 mL eppendorf tube. The mixture was incubated at 70°C for 10 min. Two hundred microliter of absolute ethanol was then added to the sample and mixed by vortexing. The mixture was then transferred to the QIAamp spin column and placed in a clean 2 mL collection tube and centrifuged at 8000 g for 1 min at room temperature. The QIAspin column was washed twice using 500 μL of washing buffers W1 and W2, respectively by spinning at 12,000 g for 1 min. The QIAamp spin column was placed in a clean 1.5 mL eppendorf tube and the DNA was eluted with 200 μL of double distilled water preheated at 70°C. Maximum DNA yield was obtained by spinning at 12,000 g for 1 min at room temperature. The bacterial DNA concentration was determined by spectrophotometer at 260 nm wave length. Five microliters of the suspended nucleic acid was used as a target DNA in the PCR amplification.
Primers Selection
A pair of oligonucleotide primers 1 and 2 (P1 and P2), designed from a highly conserved region of the uidA gene, which codes for glucuronidase specific for E. coli and Shigella was used in this study (Davies et al., 1994; Juck et al., 1996). P1 (5'-ATC ACC GTG GTG ACG CAT GTC GC-3') included 23 bases of the positive sense strand. P2 (5'-CAC CAC GAT GCC ATG TTC ATC TGC-3') included 24 bases of the complementary strand. The PCR using primer P1 and P2 will result in a 486 bp PCR product. For nested amplification, a pair of internal primers (P3 and P4) was designed from the same sequence cited above (Juck et al., 1996). P3 (5'-TAT GAA CTG TGC GTC ACA GCC-3') and P4 (5'-CAT CAG CAC GTT ATC GAA TCC-3') were used in the nested PCR to amplify a 186-bp fragment from the primary PCR. All primers were synthesized on a DNA synthesizer (Milligen/Biosearch, a division of Millipore Burlington, MA, USA) and purified using oligo-pak oligonucleotide purification columns (Glen Research Corporation, Sterling, Virginia).
Polymerase Chain Reaction (PCR)
A master mixture containing 250 μL 10X PCR buffer, 100 μL of 25 mM magnesium chloride, 12.5 μL of each dNTPs (ATP, TTP, GTP and CTP) at a concentration of 10 mM was prepared in 1.5 mL eppendorf tube. The primers were used at a concentration of 20 pg μL-1. Double distilled water was added to bring the volume of the stock buffered solution to 1.5 mL. The amount of 5.0 μL of the target DNA and 2 μL of primers were added to 42 μL of the master mixture in 0.5 mL PCR tube for each PCR amplification and mixed by vortexing. One μL of Taq DNA polymerase (Perkin Elmer) was used at a concentration of 5.0 U μL-1. All PCR amplification reactions were carried out in a final volume of 50 μL. The thermal cycling profiles were as follows: a 2-min incubation at 94°C, followed by 40 cycles of 94°C for 1 min, 57°C for 30 sec and 72°C for 45 sec. A final incubation at 72°C for 10 min was carried out to ensure complete synthesis of the expected primary 486 bp PCR products.
Thermal profiles were performed on a Techne PHC-2 thermal cycler (Techne, Princeton, NJ.). Following amplification, 10 μL from each PCR reaction containing amplified product were loaded onto gels of 1.0% SeaKem agarose (FMC Bioproduct, Rockland ME) and electrophoresed. The gels were stained with ethidium bromide and the specific 486 bp PCR products were identified following visualization under UV light.
Nested Polymerase Chain Reaction (nPCR)
Nested PCR was performed as described previously (Aradaib et al., 2005). Briefly, 5 μL of the primary amplified product, 42 μL of the master mixture and 2 μL of primers P3 and P4 at a concentration of 20 pg μL-1 were added to 0.5 mL PCR tube. 1.0 μL of Taq DNA polymerase (Perkin Elmer) was used at a concentration of 5.0 U μL-1 per reaction. All PCR amplification reactions were carried out in a final volume of 50 μL. The nested PCR amplification was performed with an initial denaturation step at 95°C for 2 min; 35 cycles at 95°C for I min, 57°C for 30 sec and 72°C for 45 sec; and a final elongation phase at 72°C for 10 min.
The products of the nested PCR were electrophoresed with 3 μL of loading buffer (0.25 g of bromphenol blue and 40 g of saccharose dissolved in 100 mL of distilled water) through a 2% agarose gel at 80 V for 45 min. Molecular size markers (Promega, Madison, Wis.) were run concurrently. The gel, stained with ethidium bromide (0.5 μg mL-1), was examined under UV light for the presence of a 186-bp nested PCR product. The ethidium bromide stained agarose gels were then photographed for documentation.
RESULTS
The described PCR-based assay allowed sensitive and specific detection of all environmental samples containing E. coli strains used in this study. The specific 486 bp PCR product was visualized on ethidium bromide-stained gel from ≥100 fg DNA of E. coli (Fig. 1). Using the internal primers (P3 and P4), the nested PCR resulted in amplification of a 186 bp PCR product. The nested 186 bp PCR product was detected from as little as 0.1 fg of E. coli DNA target (Fig. 2). Using 1 pg of E. coli DNA target extracted from purified colonies, the primary 486 bp specific PCR products was detected in eighteen E. coli strains isolated from different environmental sources (Fig. 3).
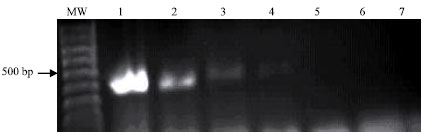 | |
| Fig. 1: | Sensitivity of the polymerase chain reaction for detection of the primary 486-bp PCR product from different DNA concentrations of E. coli. Visualization of the 486-bp specific- PCR product on ethidium bromide-stained agarose gel from 100 fg DNA. Lane MW: molecular weight marker (100 bp ladder); lanes 1-7: E. coli DNA 100, 10 and 1.0 pg, 100, 10, 1.0 and 0.1 fg, respectively |
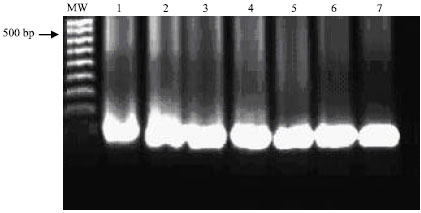 | |
| Fig. 2: | Sensitivity of the nested polymerase chain reaction (nPCR) for detection of the nested 186 bp PCR product from E. coli. Visualization of the 186-bp specific-PCR product on ethidium bromide-stained agarose gel from 100 fg DNA. Lane MW: molecular weight marker; lanes 1-7: E. coli DNA 100, 10 and 1.0 pg, 100, 10, 1.0 and 0.1 fg, respectively |
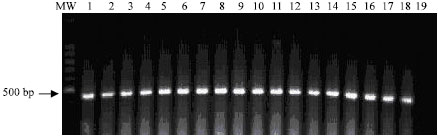 | |
| Fig. 3: | Visualization of the 486 bp specific E. coli PCR product on ethidium bromide-stained agarose gel from 1.0 pg of DNA of nineteen different E.coli strains. Lane MW: molecular weight marker; Lane 1-18: E. coli DNA; Lane 19: Negative control |
The amount of 1.0 pg bacterial DNA extracted from closely related members of enterobacteriacae including Salmonella sp., Proteus sp. and Pseudomonas; or nucleic acid-free water failed to demonstrate the primary or the nested PCR products (Fig. 4).
 | |
| Fig. 4: | Specificity of the polymerase chain reaction (PCR) for E. coli DNA. The specific 486 bp PCR product was not detected from 1.0 pg of DNA from Salmonella, Proteus and Pseudomonas or nucleic acid-free water. Lane MW: molecular weight marker; Lane 1: E.coli DNA (positive control); Lane 2-4: DNA extracts from Salmonella, Proteus and Pseudomonas sp., respectively; Lane 5: nucleic acid-free water |
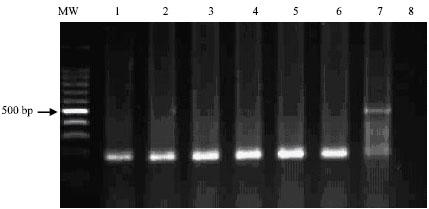 | |
| Fig. 5: | Visualization of the 186-bp specific- PCR product on ethidium bromide-stained agarose gel directly from environmental samples suspected to contain E. coli. Lane 1: Tap water; Lane 2: sewage water; Lane 3 and 4: drinking water in animal houses; Lane 5: human drinking water source; Lane 6: stagnant water; Lane 7: E. coli DNA positive control |
With nested PCR amplification, the specific PCR products were detected directly from environmental water sources suspected to be contaminated with E. coli including, sewage water, tap water, drinking water collected from animal houses, stagnant water (Fig. 5). All bacterial isolates, which were PCR positive, were also found culture positive as confirmed by conventional isolation methods.
DISCUSSION
The described nested PCR assay reproducibly and specifically detected E. coli DNA directly in contaminated environmental sample and from purified colonies of the organism and directly from environmental samples. The specific 486 bp PCR products or the nested 186 bp PCR products, visualized on ethidium bromide-stained agarose gel, were obtained from all E. coli strains. The nested PCR assay was a simple procedure that efficiently detected all E. coli strains under the stringency condition used in this study. It was easier when compared to other molecular biological techniques, many of which are lengthy and cumbersome (Aradaib et al., 2005; Darby et al., 1997; Esteban and Molleja, 1998; Garner et al., 1988; Juck et al., 1996).
The sensitivity studies indicated that the nested PCR protocol was capable of detecting the amount of 0.1 fg of total E. coli genomic DNA, which corresponds to a single organism. The nested PCR assay is almost 1,000 times more sensitive than the hybridization assays (Rath and Mach, 2000).
The specificity studies indicated that, the primary specific 486 bp PCR product was not amplified from 1.0 pg of DNA extracted from different related bacteria including Salmonella, Proteus and Pseudomonas sp., or nucleic acid free water under the same stringency condition described in this study. Temperature and time for denaturation, primer annealing and extension, enzyme and magnesium chloride concentration and number of cycles of the three temperatures per time segments were very important for maintaining sensitivity and specificity of the PCR reaction. Forty cycles of amplification were used as standard PCR procedure. The amplification procedure usually takes 2 h for the primary amplification and also 2 h for nested PCR amplification. Thus, the time required for the PCR amplification was approximately 4 h.
Excellent correlation of results from the primary and nested amplification was obtained using this PCR protocol. This finding suggests that tentative diagnosis of E. coli could be based on visualization of the primary amplified 486 bp PCR product on an ethidium bromide-stained agarose gel, since it is a simple procedure that requires only 1 h after amplification. Nested amplification is necessary to confirm the specificity of the primary amplified product and to increase the sensitivity of the PCR assay particularly, when the concentration of the bacterial DNA in the suspected environmental sample is less than 100 fg. Successful amplification was also obtained directly from environmental samples with out primary isolation in culture media as shown in Fig. 5. Lane 7 of Fig. 5, which represents E. coli DNA (positive control), contains 2 bands. The 2 bands correspond to the primary 486 bp and the nested 186 bp PCR products. This could be due to high concentration of bacterial DNA in the positive control sample, which allowed selective amplification for both primary and nested PCR products. The result of this study indicated that the described nested PCR protocol has the potential to detect E. coli in environmental samples at a very low concentration. DNA extraction was a simple procedure that takes only 15 min using the commercially available QIAamp extraction Kit. To our knowledge, there is no information available on infectivity dosage. It probably requires 5 organisms to produce the nested 186 bp PCR product by the described PCR assay. This number of bacterial organisms may represent less than one infectious unit of E. coli and that could be the reason for PCR positive but bacterial culture isolation negative results. In addition, PCR detects intact organisms, injured bacteria as well as bacterial nucleic acids. Therefore, it is not uncommon to obtain PCR-positive, but bacterial isolation negative, results from the same environmental sample (Aradaib et al., 2005; Darby et al., 1997).
The E. coli nested PCR assay described in this study can replace the need for the lengthy cumbersome bacterial isolation procedures. The time required from sample submission to interpretation of the final results was consistently 8 consecutive hours, which is affordable with in the same working day. The rapidity, sensitivity and specificity of the nested PCR assay would greatly facilitate detection of E. coli in the suspected environmental samples particularly, those associated with fecal contaminations (Edberg et al., 2000; Font et al., 1997; Fricker and Fricker, 1996). Negative and positive controls should be included in each PCR reaction to estimate the lower limit of specificity and the higher limit of sensitivity. In conclusion, the described nested PCR could be used for direct detection of E. coli strains in different environments suspected of containing the organism.
ACKNOWLEDGMENTS
This study was supported by funds from the Ministry of Science and Technology, Republic of the Sudan. We are also very grateful to Mrs. Sakeena Musa, Mrs. Lana Mustafa and Mr. Adalla M. Fadlelmula for technical assistance.
REFERENCES
- Brenner, K.P., C.C. Rankin, Y.R. Roybal, G.N. Stelma, P.V. Scarpino and A.P. Dufour, 1993. New medium for the simultaneous detection of total coliforms and Escherichia coli in water. Applied Environ. Microbiol., 59: 3534-3544.
Direct Link - Byamukama, D., F. Kansiime, R.L. Mach and A.H. Farnleitner, 2000. Determination of Escherichia coli contamination with chromocult coliform agar showed a high level of discrimination efficiency for differing fecal pollution levels in tropical waters of Kampala, Uganda. Appl. Environ. Microbiol., 66: 864-868.
CrossRefDirect Link - Carson, C.A., B.L. Shear, M.R. Ellersiock and J.D. Schnell, 2003. Comparison of ribotyping and repetitive extragenic palindromic-PCR for identification of fecal Escherichia coli from humans and animals. Applied Environ. Microbiol., 69: 1836-1839.
Direct Link - Chang, G.W., J. Brill and R. Lum, 1989. Proportion of beta-Dglucuronidase-negative Escherichia coli in human faecal samples. Applied Environ. Microbiol., 55: 335-339.
Direct Link - Darby, J.M., P. Linden, W. Pasculle and M. Saul, 1997. Utilization and diagnostic yield of blood cultures in a surgical intensive care unit. Crit. Care Med., 25: 989-994.
Direct Link - Davies, C.M., S.C. Apte, S.M. Peterson and J.L. Stauber, 1994. Plant and algal interference in bacterial beta-D-galactosidase and beta- D-glucuronidase assays. Applied Environ. Microbiol., 60: 3959-3964.
Direct Link - Edberg, S.C., E.W. Rice, M.J. Karlin and J. Allen, 2000. Escherichia coli: The best biological drinking water indicator for public health protection. J. Appl. Microbiol., 88: 106S-116S.
CrossRefDirect Link - Esteban, J., A. Molleja, R. Fernandez-Roblas and F. Soriano, 1998. Number of days required for recovery of mycobacteria from blood and other samples. J. Clin. Microbiol., 36: 1456-1457.
Direct Link - Fiksdal, L., M. Pommepuy, M.P. Caprais and I. Midttun, 1994. Monitoring of faecal pollution in coastal waters by use of rapid enzymatic techniques. Applied Environ. Microbiol., 60: 1581-1584.
PubMedDirect Link - Font, X., G. Caminal, X. Gabarell, J. Lafuente and M.T. Vicent, 1997. . On-line enzyme activity determination using the stopped technique: Application to laccase activity in pulp mill wastewater treatment. Applied Microbiol. Biotechnol., 48: 168-173.
Direct Link - Fricker, E.J. and C.P. Fricker, 1996. Alternative approaches to the detection of Escherichia coli and coliforms in water. Eur. J. Microbiol. Infect. Dis., 4: 16-20.
Direct Link - Heininger, A., M. Binder, S. Schmidt, K. Unertl, Botzenhart and G. Doring, 1999. PCR and blood culture for detection of Escherichia coli bacteremia in rats. J. Clin. Microbiol., 37: 2479-2482.
Direct Link - Juck, D.J., M. Ingram, J. Prevost, C. Coallier and C. Greer, 1996. Nested PCR protocol for the rapid detection of Escherichia coli in potable water. Can. J. Microbiol., 42: 862-866.
Direct Link - Rath, J. and R.L. Mach, 2000. Simultaneous detection and differentiation of Escherichia coli populations from environmental freshwaters by means of sequence variations in a fragment of the b-D-glucuronidase gene. Applied Environ. Microbiol., 66: 1340-1346.
Direct Link