
Laura Rodrigues Araújo
Instituto de Biotecnologia, Universidade Federal de Uberlândia, Patos de Minas, Minas Gerais, Brazil
Aulus Estevão Anjos de Deus Barbosa
Instituto de Biotecnologia, Universidade Federal de Uberlândia, Patos de Minas, Minas Gerais, Brazil
LiveDNA: 55.32664
Journal of Environmental Science and Technology
Year: 2021 | Volume: 14 | Issue: 1 | Page No.: 1-12







ABSTRACT
Background and Objective: Contamination of water bodies is one of the most impacting anthropogenic activities to the environment, therefore, it is important to understand the biological processes that allow the wastewater bioremediation. The objective of this study was to identify the main bacterial genera present in sewage treatment plants and of which are these species have genes that participate in the degradation or accumulation pathways of nitrogen, phosphorus and sulfur. Materials and Methods: Genomes of 158 bacteria species, isolated from sewage treatment plants, were analyzed in search of the following pathways: nitrification, denitrification, dissimilatory nitrate reduction, phosphorus accumulation, assimilatory sulfate reduction and dissimilatory sulfate reduction and oxidation. Results: Seventy-nine bacteria species had at least one of the complete pathways, of which 11 had 3 or more complete pathways: Acidovorax caeni, Acidovorax delafieldii, Acidovorax temperans, Burkholderia vietnamiensis, Comamonas thiooxydans, Nitrobacter vulgaris, Nitrobacter winogradskyi, Paracoccus denitrificans, Pseudomonas aeruginosa, Pseudomonas fluorescens and Thiothrixnivea. Paracoccus denitrificans stands out for having the largest number of complete pathways, possessing the genes of denitrification, dissimilatory nitrate reduction, assimilatory sulfate reduction and phosphorus accumulation processes. Conclusion: Therefore, the conclusion of this study can be used to improve the optimization of wastewater treatment processes, indicating bacteria that are more adapted for bioremediation: Paracoccus denitrificans, Thiothrixnivea and Nitrospiranitrosa.
PDF Abstract XML References Citation
Copyright: © 2021. This is an open access article distributed under the terms of the creative commons attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
How to cite this article
Laura Rodrigues Araújo and Aulus Estevão Anjos de Deus Barbosa, 2021. Identification of Bacterial Species with Nitrogen, Phosphorus and Sulfur Bioremediation Pathways in Wastewater Treatment Plants. Journal of Environmental Science and Technology, 14: 1-12.
DOI: 10.3923/jest.2021.1.12
URL: https://scialert.net/abstract/?doi=jest.2021.1.12
DOI: 10.3923/jest.2021.1.12
URL: https://scialert.net/abstract/?doi=jest.2021.1.12
INTRODUCTION
Inadequate disposal of domestic sewage in water bodies can be harmful to the environment, since it has many nutrients and a rich microbial community1,2. Depending on the concentrations, these nutrients can become pollutants, such as nitrogen, phosphorous3 and sulfur. Because they can induce eutrophication and become a risk to aquatic communities and human life. However, Nitrogen (N), Phosphorus (P) and Sulfur (S) are essential elements for all living organisms4, therefore, their excess should be treated in domestic sewage.
The fundamental reasons for treating wastewater are to prevent water source contamination and to protect public health by safeguarding water supplies against the spread of diseases5,6. Municipal wastewater is mainly comprised of water (99.9%), together with relatively small concentrations of suspended and dissolved organic and inorganic solids7. Different physical and chemical processes, such as adsorption, incineration, coagulation, precipitation and chemical oxidation, can be applied to treat wastewater6. Nevertheless, there are advantages in biological processes such as a reduction in sludge production, low operating cost and suitability for simultaneous removal of different compounds.
All biological treatment processes take advantage of the bacteria's ability to use various wastewater constituents as a source of energy for microbial metabolism and as building blocks for cell synthesis. The use of living organisms, primarily microorganisms, to degrade the environmental contaminants into less toxic forms or the process whereby organic wastes are biologically degraded, under controlled conditions, is called bioremediation7. The major microorganisms found in wastewater influents are viruses, bacteria, fungi, protozoa and nematodes5,7. However, bacteria are typically considered to be the most significant organisms consuming the organic matter in wastewater5.
New genomic, metabolic and nutritional information from bacteria in biological treatment systems could help in understanding symbiotic relationships in sewage treatment plants8-10. In addition, several aspects related to microbial communities should be considered, such as the diversity and interaction between bacteria and the environment11-13.
High throughput metagenomic sequencing enables the study of the taxonomic and functional diversity of a microbial population3,14. Comparative studies of prokaryotic genomes have revealed their complex structure and organization as well as the enormous diversity between these organisms, even among isolates of the same species15. Recent works highlight that there are about 1,700-3,600 species of bacteria in wastewater treatment plants16,17.
With an enhanced understanding of the bioremediation metabolic processes, effluent treatment plants can be enriched with specific microorganisms that enable the development of genetically modified organisms, in turn increases the efficiency of sewage treatment. Moreover, the objective of this study was to identify the main genera of bacteria present in sewage treatment plants and of which are these species have genes that participate in the degradation or accumulation pathways of nitrogen, phosphorus and sulfur. In addition, phylogenetic analyses were performed on the bacteria under this study.
MATERIALS AND METHODS
Identification of the most abundant bacterial generain water treatment plants: Identification of the dominant bacterial genera in wastewater treatment plants was observed. This study considered the genera and species identified among the most abundant and isolated in more than one study. Thousands of bacterial genera were identified in each of the works, however, in all these papers a list of the most abundant species was published. These lists were crossed to identify the most abundant species, even at different treatment plants. Bacterial genomes appearing at least two of these lists were analyzed. This study was conducted in the Bioinformatics and Molecular Analysis Laboratory, “Universidade Federal de Uberlândia”, Brazil, from February-August, 2019.
The full genome of these species were downloaded from the National Center for Biotechnology Information database and all of them were bacterial species isolated and sequenced from wastewater treatment plants, activated sludge, or sewage. Downloaded genomes were used to fabricate a database for subsequent analyses.
Identification of genes involved in nitrogen, phosphorus and sulfur metabolic pathways: Protein sequences of key genes in the metabolic pathways of nitrogen, phosphorus and sulfur were downloaded from the Kyoto Encyclopedia of Genes and Genomes database (KEGG). Genes of the following pathways were analyzed: Nitrification, denitrification, dissimilatory nitrate reduction, phosphorus accumulation18, assimilatory sulfate reduction and dissimilatory sulfate reduction and oxidation. Query protein sequences are listed in Table 1. Protein sequences19 of these genes were compared with the genome database using command line t Blastn and an e-value cutoff of 1×10–20. Venn diagrams were constructed with data generated in the blast to represent the relationships between the bacteria species that have nitrogen, phosphorus, and/or sulfur pathways genes.
| Table 1: Query genes for each bioremediation pathway | ||
| Gene name | Gene | KEGG entry |
| Nitrification | ||
| Ammonia monooxygenase subunit C | AmoC | NE2064 |
| Ammonia monooxygenase subunit A | AmoA | NE0944 |
| Ammonia monooxygenase subunit B | AmoB | NE0943 |
| Hydroxylamine dehydrogenase | Hao | NE2339 |
| Noc_0892 | ||
| Nitrate reductase/nitrite oxidoreductase, alpha subunit | NxrA | NIDE3237 |
| N297_4001 | ||
| Nitrate reductase/nitrite oxidoreductase, beta subunit | NxrB | b1225 |
| SCV20265_1123 | ||
| Denitrification | ||
| Nitrate reductase gamma subunit | NarI | N296_3998 |
| Nitrate reductase/nitrite oxidoreductase, alpha subunit | NarG | BN889_04303 |
| AK36_5148 | ||
| Nitrate reductase/nitrite oxidoreductase, beta subunit | NarH | b1225 |
| UIB01_03910 | ||
| Periplasmic nitrate reductase | NapA | b2206 |
| UIB01_15470 | ||
| Cytochrome c-type protein | NapB | PA14_49260 CAP2UW1_3909 |
| Nitrite reductase | NirK | BMA10229_0703 |
| Neut_1403 | ||
| Nitrite reductase/ hydroxylamine reductase | NirS | PSE_0898 |
| Nitric oxide reductase subunit B | NorB | NE2004 |
| BMA0633 | ||
| Nitric oxide reductase subunit C | NorC | Neut_0521 |
| Nitrous-oxide reductase | NosZ | PA14_20200 |
| DNRA | ||
| Nitrite reductase (NADH) large subunit | NirB | b3365 |
| PSEEN1418 | ||
| Nitrite reductase (NADH) small subunit | NirD | Ent638_3794 |
| Pden_4451 | ||
| Nitrite reductase (cytochrome c-552) | NrfA | b4070 |
| Cj1357c | ||
| Cytochrome c nitrite reductase small subunit | NrfH | HCBAA847_0636 |
| Cj1358c | ||
| Desgi_2941 | ||
| Assimilatory sulfate reduction | ||
| 3'-phosphoadenosine 5'-phosphosulfate synthase | PAPSS | sce5751 |
| Sulfate adenylyltransferase | Sat | Tbd_0210 |
| UZ73_02605 | ||
| Sulfate adenylyltransferase subunit 1 | CysN | b2751 |
| ECL_04101 | ||
| KPN_03113 | ||
| Sulfate adenylyltransferase subunit 1 | CysD | ECL_04100 |
| KPN_03114 | ||
| Adenylyl sulfate kinase | CysC | b2750 |
| ECL_04099 | ||
| Phosphoadenosine phosphosulfate reductase | CysH | ECL_04104 |
| PA1756 | ||
| Sulfite reductase (NADPH) hemoprotein beta-component | CysI | ECL_04105 |
| CtCNB1_3170 | ||
| Sulfite reductase (NADPH) flavoprotein alpha-component | CysJ | CtCNB1_3038 |
| ENC_30120 | ||
| Sulfite reductase (ferredoxin) | Sir | Abu_2013 |
| Clopa_4350 | ||
| DSR | ||
| Adenylyl sulfate reductase, subunit A | AprA | Tbd_0872 |
| Desaf_0101 | ||
| Clopa_4347 | ||
| EUBREC_2472 | ||
| Adenylyl sulfate reductase, subunit B | AprB | Tbd_0873 |
| EUBREC_2471 | ||
| Desaf_0100 | ||
| Dissimilatory sulfite reductase alpha subunit | DsrA | Tbd_1309 |
| Desaf_1370 | ||
| Desca_2666 | ||
| Dissimilatory sulfite reductase beta subunit | DsrB | Desca_2665 |
| Tbd_2484 | ||
| Desaf_1371 | ||
| Pho | ||
| Acetate kinase | AckA | b2296 |
| Ent638_2840 | ||
| Phosphate acetyltransferase | Pta | PST_0690 |
| BMAA0121 | ||
| Acetyl-CoA synthetase | Acs | b4069 |
| AKI40_4606 | ||
| Polyhydroxyalkanoate synthase | PhaC | PST_0683 |
| O23A_p1564 | ||
| Poly(3-hydroxybutyrate) depolymerase | PhaZ | AC233_04595 |
| DNRA: Dissimilatory nitrate reduction, DSR: Dissimilatory sulfate reduction and oxidation, Pho: Phosphorus accumulation | ||
Phylogenetic analysis: Phylogenetic analysis was performed on bacterial species that showed at least one of the complete pathways listed in Table 1. Thus, 16S rRNA gene was used to compare all the analyzed bacteria. Sequence alignments were performed using CLUSTALW in the BioEdit Sequence Alignment Editor20. Phylogenetic tree construction was performed by the neighbor-joining method using the software MEGA-X21. Robustness was paramount and assessed by bootstrap analysis based on 1,000 repetitions.
Detailed analysis of the bacterial species with genes of several pathways: Bacterial species presenting three or more complete pathways for degradation and/or accumulation of nitrogen, phosphorus and sulfur were subjected to a detailed analysis. Reverse BLASTp and InterProScan were used to identify and confirm the best blast hits, which were then analyzed with Blast2GO 522 basic software.
RESULTS
Identification of the main bacterial genera in wastewater treatment plants: Identification of the dominant bacterial genera in wastewater treatment plants was based on previous studies data that performed metagenomic analyses in 8 different wastewater treatment plants (Table 2). Studies that conferred information about major genera and species of bacteria were considered as well as the proportion of each group within a sewage treatment plant.
In this study, the genome of 158 bacteria species belonging to 80 genera were scrutinized (Table 4). Genera present among the most abundant and identified in the largest number of analyzed wastewater treatment plants were listed in Table 3.
These 158 species (Table 4) were classified into 22 bacterial classes and one un classified: Acidimicrobiia, Actinobacteria, Alphaproteobacteria, Anaerolineae, Bacilli, Bacteroidia, Betaproteobacteria, Clostridia, Coriobacteriia, Deltaproteobacteria, Epsilonproteobacteria, Flavobacteriia, Gammaproteobacteria, Gemmatimonadetes, Negativicutes, Nitrospira, Oligoflexia, Rubrobacteria, Saprospiria, Sphingobacteriia, Spirochaetia and Synergistia.
Identification of genes involved in nitrogen, phosphorus and sulfur metabolic pathways and phylogenetic analysis: After analysis of the 158 bacterial genomes, 79 species conferred at least one of the complete pathways (Table 4). A Venn diagram was constructed to enhance visualization and analyze the relationship between78 bacterial species and pathways. Figure 1 correlates the pathways of phosphorus accumulation, assimilatory sulfate reduction, denitrification, dissimilatory nitrate reduction and dissimilatory sulfate reduction and oxidation. Nitrification pathway were not added to Fig. 1 because it were present in a two bacteria species (Table 4), this made it impossible to construct the Venn diagram with all pathways.
| Table 2: Scientific references of main bacterial genera in wastewater treatment plants | |||
| Country | pH | Temperature (°C) | References |
| China | 7.3-7.8 | 35 | M.C. Macey et al.26 |
| Belgium | 6.77-7.76 | 8.3-21.1 | K. Meerbergen et al.16 |
| China | 7 | 34-36 | Q. Ma et al.39 |
| China | Not show | Not show | Y. Yang et al.40 |
| China | Not show | Not show | L. Cai et al.1 |
| China | 6.4-7.3 | Not show | Q. Ma et al.39 |
| China | 6.75-7.5 | 8.5-13.5 | Y. Yang et al.40 |
| China | Not show | 31-32 | Q. Huang et al.41 |
| Table 3: Most abundant genera and the number of identified wastewater treatment plants | ||
| Genus | Class | Number of identifications |
| Clostridium | Clostridia | 7 |
| Nitrospira | Nitrospira | 6 |
| Bacteroides | Bacteroidia | 5 |
| Pseudomonas | Gammaproteobacteria | 5 |
| Thauera | Betaproteobacteria | 5 |
| Acidovorax | Betaproteobacteria | 4 |
| Dechloromonas | Betaproteobacteria | 4 |
| Dokdonella | Gammaproteobacteria | 4 |
| Mycobacterium | Actinobacteria | 4 |
| Streptococcus | Bacilli | 4 |
| Arcobacter | Epsilonproteobacteria | 3 |
| Bacillus | Bacilli | 3 |
| Bifidobacterium | Actinobacteria | 3 |
| Flavobacterium | Flavobacteriia | 3 |
| Lactobacillus | Bacilli | 3 |
| Paracoccus | Alphaproteobacteria | 3 |
| Rhodobacter | Alphaproteobacteria | 3 |
| Treponema | Spirochaetia | 3 |
Phylogenetic analysis was conducted with all bacteria that presented at least one of the complete pathways (Fig. 2). These 79 bacteria accounted for half of the initial sample and were divided into 9 classes and for majority of them were classified in the phylum Proteobacteria (86%). In addition, the analyses indicated that this phylum has the bacteria species with more genes for bioremediation. Initially, 83 Proteobacteria species were analyzed and 63of them (75.9%) had at least one of the nitrogen’s, sulfur or phosphorus pathways.
Nitrification was the least observed pathway, with only two bacteria species presenting the complete pathway: Candidatus Nitrospiranitrificans and Candidatus Nitrospiranitrosa (Fig. 2). In addition, all Nitrospira analyzed are among the most abundant in wastewater treatment plants (Table 3). Otherwise, denitrification was most common and was observed in 9 bacteria species (Fig. 1).
Twelve species conferred with complete dissimilatory sulfate reduction and oxidation pathway (DSR) but only Thiothrixnivea, from class Gammaproteobacteria, also has the complete pathways of assimilatory sulphate reduction and dissimilatory nitrate reduction (Fig. 1). Only Eight species of these bacteria belong to the class of Deltaproteobacteria and three species are classified in the Clostridia class (Fig. 2).
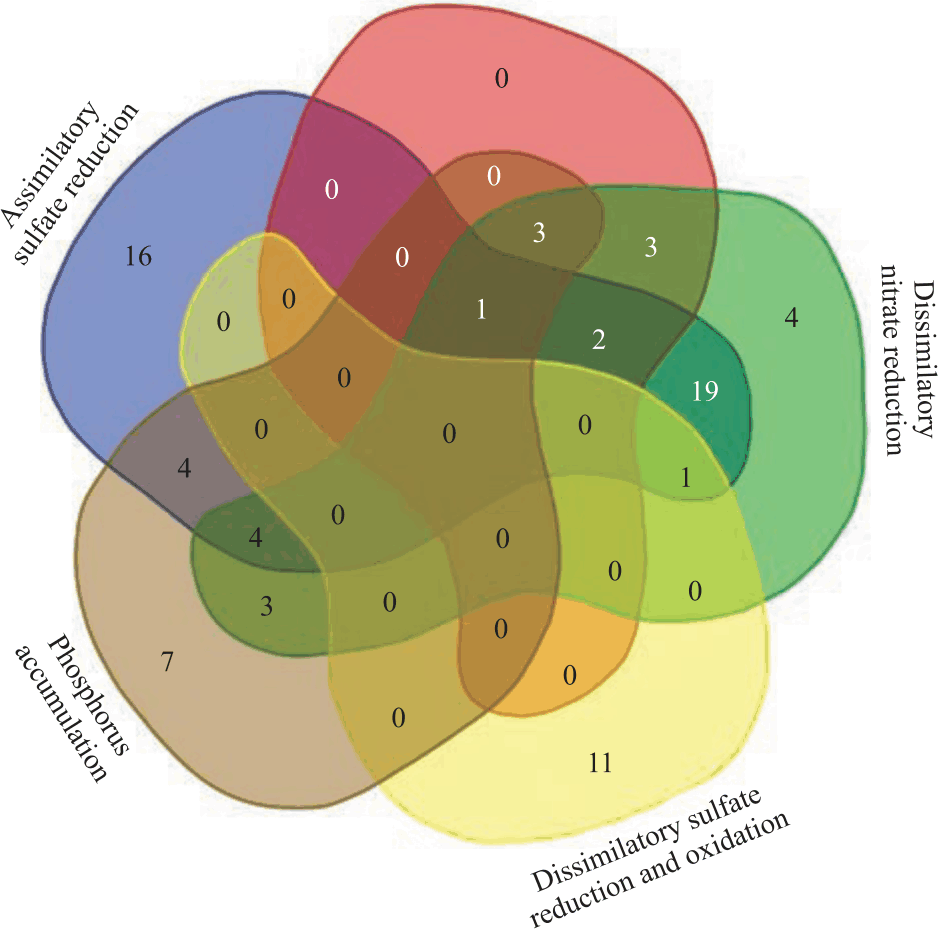 |
| Fig. 1: | Venn diagram correlating 78 bacterial species that presented all genes of one or more pathways |
Forty species of bacteria include at least two of the analyzed pathways (Table 4). Blast experiments showed that 47 bacterial species possess the assimilatory sulfate reduction pathway genes and the other 40 possess dissimilatory nitrate reduction to ammonia (DNRA) pathway genes-also known as nitrate/nitrite ammonification. These two pathways are the most observed in the bacterial genomes. Among these species, 27granted all the genes related to both pathways, assimilatory sulfate reduction and DNRA (Fig. 1). From these 27bacteria species mentioned above, 22of them belong to the class Gammaproteobacteria and the others belong to the categories Alphaproteobacteria and Betaproteobacteria (all belonging to the phylum Proteobacteria) (Fig. 2).
Bacterial species with genes of three or more pathways: Forty species exhibited all genes with more than one pathway (Fig. 1) but 11 of these bacteria had three or more complete pathways.
| Table 4: Bacteria list indicating complete bioremediation pathways presence | ||||||||
| No. | ID - NCBI | Specie | Nitrification | Denitrification | DNRA | ASR | DSR | Phosphor |
| 1 | NZ_CP014692 | Acetobacter aceti | X | |||||
| 2 | NZ_CP020917 | Achromobacter denitrificans | X | |||||
| 3 | NZ_CYIG01000001.1 | Acidovorax caeni | X | X | X | |||
| 4 | NZ_ACQT01000638.1 | Acidovorax delafieldii | X | X | X | |||
| 5 | NZ_JXYQ01000001.1 | Acidovorax temperans | X | X | X | |||
| 6 | NC_014259 | Acinetobacter oleivorans | ||||||
| 7 | NC_008570.1 | Aeromonas hydrophila | X | X | ||||
| 8 | NZ_CP007567.1 | Aeromonas media | X | X | ||||
| 9 | NZ_CP013119.1 | Alcaligenes faecalis | X | X | ||||
| 10 | NZ_JH370371.1 | Alistipes indistinctus | ||||||
| 11 | NZ_JRGF01000001.1 | Alistipes inops | ||||||
| 12 | NZ_DS499581.1 | Alistipes putredinis | ||||||
| 13 | NC_014011 | Aminobacterium colombiense | ||||||
| 14 | NZ_JAFZ01000001.1 | Aminobacterium mobile | ||||||
| 15 | NC_013171.1 | Anaerococcus prevotii | ||||||
| 16 | NC_014960.1 | Anaerolinea thermophila | ||||||
| 17 | NC_009850.1 | Arcobacter butzleri | ||||||
| 18 | NZ_NWVW01000010.1 | Arcobacter canalis | ||||||
| 19 | NZ_JARS01000021.1 | Arcobacter faecis | ||||||
| 20 | NC_011886.1 | Arthrobacter chlorophenolicus | ||||||
| 21 | NZ_CP018863.1 | Arthrobacter crystallopoietes | ||||||
| 22 | NZ_CP007514.1 | Arthrobacter radiotolerans | ||||||
| 23 | NC_006274 | Bacillus cereus | ||||||
| 24 | NC_000964.3 | Bacillus subtilis | ||||||
| 25 | NC_022873.1 | Bacillus thuringiensis | ||||||
| 26 | NZ_CP011531 | Bacteroides dorei | ||||||
| 27 | NZ_AKBZ01000001.1 | Bacteroides finegoldii | ||||||
| 28 | NC_006347.1 | Bacteroides fragilis | ||||||
| 29 | NZ_CP012938.1 | Bacteroides ovatus | ||||||
| 30 | NC_004663.1 | Bacteroides thetaiotaomicron | ||||||
| 31 | NC_009614.1 | Bacteroides vulgatus | ||||||
| 32 | NC_021017.1 | Bacteroides xylanisolvens | ||||||
| 33 | NC_005363 | Bdellovibrio bacteriovorus | ||||||
| 34 | NZ_CP012373.1 | Beggiatoa leptomitoformis | X | |||||
| 35 | NC_008618.1 | Bifidobacterium adolescentis | ||||||
| 36 | NC_012815 | Bifidobacterium animalis | ||||||
| 37 | NC_014638.1 | Bifidobacterium bifidum | ||||||
| 38 | NZ_AUAO01000001.1 | Brevundimonas aveniformis | ||||||
| 39 | NZ_JNIX01000001.1 | Brevundimonas bacteroides | ||||||
| 40 | NZ_CP009323.1 | Burkholderia gladioli | X | |||||
| 41 | NZ_CP013433.1 | Burkholderia vietnamiensis | X | X | X | |||
| 42 | FLQX01000001.1 | Candidatus Accumulibacter Aalbogensi | X | |||||
| 43 | NC_013194 | Candidatus Accumulibacter phosphatis | X | |||||
| 44 | NC_020449.1 | Candidatus Cloacamonas acidaminovorans | ||||||
| 45 | NZ_HG422565.1 | Candidatus Microthrix parvicella | ||||||
| 46 | NC_014355 | Candidatus Nitrospira defluvii | X | |||||
| 47 | NZ_CZPZ01000001.1 | Candidatus Nitrospira nitrificans | X | |||||
| 48 | NZ_CZQA01000001.1 | Candidatus Nitrospira nitrosa | X | X | ||||
| 49 | NZ_JARQ01000001.1 | Chryseobacterium hispalense | ||||||
| 50 | NZ_LFNG01000001.1 | Chryseobacterium koreense | ||||||
| 51 | NZ_CP007557 | Citrobacter freundii | X | X | ||||
| 52 | NZ_CP019986 | Citrobacter werkmanii | X | X | ||||
| 53 | NZ_GG730308.1 | Citrobacter youngae | X | X | ||||
| 54 | NZ_CP013252.1 | Clostridium butyricum | ||||||
| 55 | NZ_CP017603 | Clostridium formicaceticum | ||||||
| 56 | NZ_ACXX02000031.1 | Clostridium papyrosolvens | ||||||
| 57 | NC_021182.1 | Clostridium pasteurianum | ||||||
| 58 | NZ_CP024160.1 | Collinsella aerofaciens | ||||||
| 59 | NZ_CP016603.1 | Comamonas aquatica | X | X | ||||
| 60 | NZ_AXVM01000001.1 | Comamonas badia | X | |||||
| 61 | NZ_BBJX01000034.1 | Comamonas granuli | X | |||||
| 62 | NZ_CP020121.1 | Comamonas kerstersii | X | X | ||||
| 63 | NZ_CP006704 | Comamonas testosteroni | X | |||||
| 64 | NZ_CYHD01000001.1 | Comamonas thiooxydans | X | X | X | |||
| 65 | NC_004369.1 | Corynebacterium efficiens | ||||||
| 66 | NC_003450.3 | Corynebacterium glutamicum | ||||||
| 67 | NC_007298.1 | Dechloromonas aromatica | X | X | ||||
| 68 | NC_013173 | Desulfomicrobium baculatum | X | |||||
| 69 | NZ_AUAR01000001.1 | Desulfomicrobium escambiense | X | |||||
| 70 | NC_013216 | Desulfotomaculum acetoxidans | X | |||||
| 71 | NC_021184.1 | Desulfotomaculum gibsoniae | X | |||||
| 72 | NC_015565 | Desulfotomaculum nigrificans | X | |||||
| 73 | NC_016629.1 | Desulfovibrio africanus | X | |||||
| 74 | NZ_KE383875.1 | Desulfovibrio aminophilus | X | |||||
| 75 | NZ_KE386885.1 | Desulfovibrio putealis | X | |||||
| 76 | NC_012881 | Desulfovibrio salexigens | X | |||||
| 77 | NC_002937 | Desulfovibrio vulgaris | X | |||||
| 78 | NZ_CP017037.1 | Dialister pneumosintes | ||||||
| 79 | NZ_CP015249.1 | Dokdonella koreensis | X | |||||
| 80 | CP003026.1 | Enterobacter asburiae | X | X | ||||
| 81 | CP025225.1 | Enterobacter cancerogenus | X | X | ||||
| 82 | NC_014121.1 | Enterobacter cloacae | X | X | ||||
| 83 | NC_020995.1 | Enterococcus casseliflavus | ||||||
| 84 | NC_004668 | Enterococcus faecalis | ||||||
| 85 | NZ_CP016625 | Escherichia coli | X | X | ||||
| 86 | NC_012778.1 | Eubacterium eligens | ||||||
| 87 | NC_012781.1 | Eubacterium rectale | ||||||
| 88 | NZ_GG688422.1 | Eubacterium saphenum | ||||||
| 89 | NZ_KB907512.1 | Eubacterium siraeum | ||||||
| 90 | NZ_DS264288.1 | Eubacterium ventriosum | ||||||
| 91 | NZ_CP030777.1 | Faecalibacterium prausnitzii | ||||||
| 92 | NC_009441.1 | Flavobacterium johnsoniae | ||||||
| 93 | NC_015321 | Fluviicola taffensis | ||||||
| 94 | NC_012489.1 | Gemmatimonas aurantiaca | ||||||
| 95 | NZ_CP011454.1 | Gemmatimonas phototrophica | ||||||
| 96 | NZ_CP014963.1 | Geobacter anodireducens | ||||||
| 97 | NC_002939 | Geobacter sulfurreducens | ||||||
| 98 | CP009706.1 | Hafnia alvei | X | X | ||||
| 99 | NZ_LXET01000001.1 | Hafnia paralvei | X | X | ||||
| 100 | NC_015510.1 | Haliscomenobacter hydrossis | ||||||
| 101 | NZ_CP011636.1 | Klebsiella oxytoca | X | X | ||||
| 102 | NZ_CP020847 | Klebsiella pneumoniae | X | X | ||||
| 103 | NZ_CP016766 | Lactobacillus agilis | ||||||
| 104 | NC_004342.2 | Leptospira interrogans | ||||||
| 105 | NC_012803.1 | Micrococcus luteus | ||||||
| 106 | NZ_FPCG01000031.1 | Micrococcus terreus | ||||||
| 107 | NZ_CP007220 | Mycobacterium chelonae | X | |||||
| 108 | NC_007964 | Nitrobacter hamburgensis | X | X | ||||
| 109 | NZ_MWPQ01000040.1 | Nitrobacter vulgaris | X | X | X | |||
| 110 | NC_007406 | Nitrobacter winogradskyi | X | X | X | |||
| 111 | NC_004757 | Nitrosomonas europaea | ||||||
| 112 | NC_008344.1 | Nitrosomonas eutropha | X | |||||
| 113 | NZ_FODO01000081.1 | Nitrosomonas oligotropha | X | |||||
| 114 | NZ_CP021106.3 | Nitrosospira lacus | ||||||
| 115 | NC_007614 | Nitrosospira multiformis | X | |||||
| 116 | NZ_LT828648.1 | Nitrospira japonica | X | |||||
| 117 | NZ_CP011801.1 | Nitrospira moscoviensis | ||||||
| 118 | NZ_FUYQ01000044.1 | Parabacteroides chartae | X | |||||
| 119 | NC_009615 | Parabacteroides distasonis | ||||||
| 120 | NZ_KE159513.1 | Parabacteroides goldsteinii | ||||||
| 121 | NZ_JH976465.1 | Parabacteroides johnsonii | ||||||
| 122 | NZ_JH976452.1 | Parabacteroides merdae | ||||||
| 123 | NC_008686.1 | Paracoccus denitrificans | X | X | X | X | ||
| 124 | CP025430.1 | Paracoccus zhejiangensis | X | X | ||||
| 125 | NC_013061.1 | Pedobacter heparinus | ||||||
| 126 | NC_007498 | Pelobacter carbinolicus | ||||||
| 127 | NC_008609.1 | Pelobacter propionicus | ||||||
| 128 | CZAM01000001.1 | Prevotella copri | ||||||
| 129 | NZ_CP021852 | Proteus mirabilis | X | X | ||||
| 130 | GG662004.1 | Proteus penneri | X | |||||
| 131 | NC_002516.2 | Pseudomonas aeruginosa | X | X | X | |||
| 132 | NC_016830 | Pseudomonas fluorescens | X | X | X | |||
| 133 | NC_002947.4 | Pseudomonas putida | ||||||
| 134 | NC_014034.1 | Rhodobacter capsulatus | X | |||||
| 135 | NC_009428.1 | Rhodobacter sphaeroides | X | |||||
| 136 | NC_008268.1 | Rhodococcus jostii | X | |||||
| 137 | NC_003197.2 | Salmonella enterica | X | X | ||||
| 138 | NZ_CP011642 | Serratia marcescens | X | X | ||||
| 139 | NZ_AP017655.1 | Sphingobium cloacae | X | X | ||||
| 140 | NZ_CP012900.1 | Stenotrophomonas acidaminiphila | X | X | ||||
| 141 | NC_016826 | Streptococcus infantarius | ||||||
| 142 | NZ_CP007201.1 | Sulfurospirillum multivorans | X | |||||
| 143 | NC_008554.1 | Syntrophobacter fumaroxidans | X | |||||
| 144 | NZ_BBCE01000001.1 | Syntrophomonas palmitatica | ||||||
| 145 | NC_008346.1 | Syntrophomonas wolfei | ||||||
| 146 | NZ_CGIH01000001.1 | Syntrophomonas zehnderi | ||||||
| 147 | NC_007759.1 | Syntrophus aciditrophicus | ||||||
| 148 | NZ_CP028339.1 | Thauera aromatica | X | X | ||||
| 149 | NC_007404 | Thiobacillus denitrificans | X | X | ||||
| 150 | NZ_CP020046 | Thiomonas intermedia | X | |||||
| 151 | NZ_KB904746.1 | Thiothrix flexilis | X | |||||
| 152 | NZ_JH651381.1 | Thiothrix nivea | X | X | X | |||
| 153 | NC_015732.1 | Treponema caldarium | ||||||
| 154 | NC_014158.1 | Tsukamurella paurometabola | X | |||||
| 155 | NC_009456.1 | Vibrio cholerae | X | |||||
| 156 | NZ_JMCG01000001.1 | Vibrio navarrensis | X | X | ||||
| 157 | NZ_CQBU01000001.1 | Yersinia bercovieri | X | X | ||||
| 158 | CP009364.1 | Yersinia frederikseni | X | X | ||||
| Bacteria shaded in gray do not have any complete pathway | ||||||||
Paracoccus denitrificans stands out for having the greatest number of complete pathways, possessing the genes of denitrification, dissimilatory nitrate reduction, assimilatory sulfate reduction and phosphorus accumulation processes (Fig. 1).
Thiothrixnivea was the only species that possessed the genes necessary to complete the two routes of sulfur, both assimilatory (ASR) and dissimilatory sulfate reduction (DSR) (Fig. 1).
Burkholderia vietnamiensis, Comamonas thiooxydans, Nitrobacter vulgaris and Nitrobacter winogradskyi have genes that act on the same pathways, i.e., dissimilatory nitrate reduction, assimilatory sulfate reduction and phosphorus accumulation (Fig. 1). Burkholderia vietnamiensis and Comamonas thiooxydans belong to the Betaproteobacteria class, while Nitrobacter vulgaris and Nitrobacter winogradskyi belong to the Alphaproteobacteria class (Fig. 2).
 |
| Fig. 2: | 16S rRNA phylogeny of bacteria presented at least one of the complete pathways Class level lineages are indicated on the blue lines |
DISCUSSION
In this study, 158 bacterial genomes were analyzed in search of genes that act in six bioremediation processes. Half of the analyzed species (79) have at least one of the complete pathways (Table 4) and 18 of them have 3 or more analyzed pathways (Table 3), which indicate a better metabolic capacity of these species.
Gammaproteo bacteria was the most leading class, with 18.9% of analyzed species, followed by Betaproteobacteria (15.1%), Clostridia (10.7%), Bacteroidia (10.2%), Deltaproteobacteria (8.2%), Actinobacteria (7.5%) and Alphaproteobacteria (6.9%), with predominant species belonging to the phylum Proteobacteria (52.5%). This result was similar with other studies23-26, indicating that this study sampling with the most common bacteria in different sewage treatment plants is significant.
Clostridium was the most identified genus in different wastewater treatment plants analyzed (Table 3) and these bacteria are found in different environments and inhabiting the intestines of several animal species27,28. The second most identified genus was Nitrospire, that are found in many environments and are responsible for nitrification processes29. Remarkably, all bacteria listed in Table 3 belong to the phylum Proteobacteria. These species presented genes that enable them to act in cycles of the three nutrients studied (nitrogen, phosphorus and sulfur). Because of this, it is possible that Proteobacteria is the predominant phylum in practically all sewage treatment plants analyzed30,31.
Nitrification was the least observed pathway, with two Nitrospira bacteria presenting the complete pathway (Fig. 2). Separation of nitrification into two steps led to a cross-feeding interaction between different species of bacteria. On the other hand, those that could catalyze the complete nitrification pathway had growth advantages over the others32.
Dissimilatory sulfate reduction and oxidation pathway (DSR) are present in 12 bacteria species (Fig. 1). SRB controlled application in wastewater treatment carry several advantages: promotes pathogen and heavy metal removal, reduction of sludge disposal and perform a pre-treatment before anaerobic digestion that results in higher biogas yields33.
Assimilatory Sulfate Reduction (ASR) and Dissimilatory Nitrate Reduction (DNRA) are the pathways present in a greater number of bacteria (Fig. 1). Recent studies have demonstrated that the nitrogen cycle is also tightly linked to other biogeochemical cycles, such as the sulfur cycle34,35 and suggest that biogenic sulfide induces DNRA with coproduction of ammonium and nitrite36.
Denitrification pathway was observed in 9 bacteria species and these bacteria also have genes from other pathways (Fig. 1). Sulfate reduction can indirectly stimulate P release and when sulfate is reduced to sulfide, this molecule can bind its self to Fe(II), leading to more P availability37. Curiously, four bacteria species were identified with genes for performing denitrification and phosphorus accumulation (P. denitrificans and three bacteria of Acidovorax genus) (Fig. 1). Therefore, this combination of genes should make these species efficient nitrate and phosphorus removers.
The bacterium with the greatest capacity for bioremediation is Paracoccus denitrificans possessing the genes of 4 pathways: denitrification, dissimilatory nitrate reduction, assimilatory sulfate reduction and phosphorus accumulation processes (Fig. 1). It is a nonmotile coccoid soil organism and is taxonomically part of the Rhodobacteraceae family from a subdivision of the phylum Proteobacteria38. P. denitrificans can live in oxic and anoxic environments in response to environmental changes, such as oxygen and nitrogenous oxide concentration.
Another interesting bacterium in the bioremediation process is Thiothrixnivea, having all genes for assimilatory (ASR) and dissimilatory sulfate reduction (DSR) (Fig. 1). ASR is characterized by sulfate reduction in small amounts required for the synthesis of cellular material, whereas DSR is described as the sulfate reduction in great excess of nutritional requirements, producing massive amounts of sulfide32.
The main limitation of this study was to have analyzed a fraction of the species in a wastewater treatment plant, which has 1700-3600 species16,17. However, the 158 analyzed species are quite representative, being the most abundant in several wastewater treatment plants. Identification of the main species described in the study, such as Paracoccus denitrificans, Thiothrixnivea and Nitrospiranitrosa, would allow to evaluate in vitro the metabolic capacities of these species in the wastewater bioremediation.
Therefore, the results of this observation could be used to increase the sewage treatment efficiency, indicating/ allocating the most appropriate bacterial species in degradation of nitrogen, phosphorus and sulfur compounds. Additionally, this study could be used in the development of more coherent genetically modified organisms in wastewater bioremediation.
CONCLUSION
Knowledge of the bacterial community composition and how it interacts inside the wastewater treatment plants are essential for better designed bioremediation strategies. Bacteria having genes for the pathways studied here can be introduced into a sewage treatment plant to increase organic matter degradation, for example, P. denitrificans, T. and N. nitrosa. A combination of these three bacteria would have all the genes analyzed in this study.
SIGNIFICANCE STATEMENT
This study identified the main bacterial species that perform wastewater bioremediation process and these results can be used to improve bioremediation processes. The bacteria indicated in the study can be added to treatment plants in a selective enrichment method, increasing nitrogen, sulfur and phosphorus compounds bioremediation.
ACKNOWLEDGMENT
This study was supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior CAPES-Brazil (CAPES). The authors are grateful to LBAM/UFU for providing the infrastructure for the development of this study.
REFERENCES
- Cai, L., F. Ju and T. Zhang, 2014. Tracking human sewage microbiome in a municipal wastewater treatment plant. Appl. Microbiol. Biotechnol., 98: 3317-3326.
CrossRefDirect Link - Turki, Y., I. Mehri, R. Lajnef, A.B. Rejab and A. Khessairi et al., 2017. Biofilms in bioremediation and wastewater treatment: Characterization of bacterial community structure and diversity during seasons in municipal wastewater treatment process. Environ. Sci. Pollut. Res. Int., 24: 3519-3530.
CrossRefDirect Link - Ye, Y., H.H. Ngo, W. Guo, Y. Liu and X. Zhang, 2016. Insight into biological phosphate recovery from sewage. Bioresour. Technol., 218: 874-881.
CrossRefDirect Link - Gerardi, M.H., 2006. Wastewater Bacteria. 1st Edn., John Wiley and Sons, New York, ISBN-13: 978-0471206910, Pages: 272.
Direct Link - Akpor, O.B. and M. Muchie, 2010. Bioremediation of polluted wastewater influent: Phosphorus and nitrogen removal. Sci. Res. Essays, 5: 3222-3230.
CrossRefDirect Link - Khan, M.Z., P.K. Mondal and S. Sabir, 2013. Aerobic granulation for wastewater bioremediation: A review. Can. J. Chem. Eng., 91: 1045-1058.
CrossRefDirect Link - Amin, A., A.T.R. Naik, M. Azhar and H. Nayak, 2013. Bioremediation of different waste waters-a review. Cont. J. Fish. Aquat. Sci., 7: 7-17.
CrossRefDirect Link - Sanz, J.L. and T. Kochling, 2007. Molecular biology techniques used in wastewater treatment: An overview. Process Biochem., 42: 119-133.
CrossRefDirect Link - Yang, Y., K. Yu, Y. Xia, F.T.K. Lau and D.T.W. Tang et al., 2014. Metagenomic analysis of sludge from full-scale anaerobic digesters operated in municipal wastewater treatment plants. Appl. Microbiol. Biotechnol., 98: 5709-5718.
CrossRefDirect Link - Jałowiecki, Ł., J.M. Chojniak, E. Dorgeloh, B. Hegedusova, H. Ejhed, J. Magnér, G.A. Płaza, 2016. Microbial Community Profiles in Wastewaters from Onsite Wastewater Treatment Systems Technology. PLoS ONE, Vol. 11.
CrossRefDirect Link - Vanwonterghem, I., P.D. Jensen, D.P. Ho, D.J. Batstone and G.W. Tyson, 2014. Linking microbial community structure, interactions and function in anaerobic digesters using new molecular techniques. Curr. Opin. Biotechnol., 27: 55-64.
CrossRefDirect Link - Ju, F. and T. Zhang, 2015. Bacterial assembly and temporal dynamics in activated sludge of a full-scale municipal wastewater treatment plant. ISME J., 9: 683-695.
CrossRefDirect Link - Cydzik-Kwiatkowska, A. and M. Zielinska, 2016. Bacterial communities in full-scale wastewater treatment systems. World J. Microbiol. Biotechnol., 32: 66-69.
CrossRefDirect Link - Garrido-Cardenas, J.A. and F. Manzano-Agugliaro, 2017. The metagenomics worldwide research. Curr. Genet., 63: 819-829.
CrossRefDirect Link - Touchon, M. and E.P.C. Rocha, 2016. Coevolution of the organization and structure of prokaryotic genomes. Cold Spring Harbor Perspect. Biol., Vol. 8.
CrossRefDirect Link - Meerbergen, K., M.V. Geel, M. Waud, K.A. Willems and R. Dewil et al., 2016. Assessing the composition of microbial communities in textile wastewater treatment plants in comparison with municipal wastewater treatment plants. Microbiologyopen, Vol. 6.
CrossRefDirect Link - Zhang, B., X. Xu and L. Zhu, 2018. Activated sludge bacterial communities of typical wastewater treatment plants: distinct genera identification and metabolic potential differential analysis. AMB Expr., Vol. 8.
CrossRefDirect Link - Albertsen, M., S.J. McIlroy, M. Stokholm-Bjerregaard, S.M. Karst and P.H. Nielsen, 2016. “Candidatus propionivibrio aalborgensis”: A novel glycogen accumulating organism abundant in full-scale enhanced biological phosphorus removal plants. Front. Microbiol., Vol. 7.
CrossRefDirect Link - Altschul, S.F., W. Gish, W. Miller, E.W. Myers and D.J. Lipman, 1990. Basic local alignment search tool. J. Mol. Biol., 215: 403-410.
CrossRefPubMedDirect Link - Hall, T.A., 1999. BioEdit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acid Symp. Ser., 41: 95-98.
Direct Link - Kumar, S., G. Stecher, M. Li, C. Knyaz and K. Tamura, 2018. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol., 35: 1547-1549.
CrossRefDirect Link - Gotz, S., J.M. Garcia-Gomez, J. Terol, T.D. Williams and S.H. Nagaraj et al., 2008. High-throughput functional annotation and data mining with the blast2GO suite. Nucleic Acids Res., 36: 3420-3435.
CrossRefDirect Link - Xu, S., J. Yao, M. Ainiwaer, Y. Hong and Y. Zhang, 2018. Analysis of bacterial community structure of activated sludge from wastewater treatment plants in winter. Biomed. Res. Int., Vol. 2018.
CrossRefDirect Link - Tao, W., X.X. Zhang, F. Zhao, K. Huang and H. Ma, Z. Wang, L. Ye and H. Ren, 2016. High levels of antibiotic resistance genes and their correlations with bacterial community and mobile genetic elements in pharmaceutical wastewater treatment bioreactors. PLoS ONE.
CrossRefDirect Link - Guo, J., Y. Peng, B.J. Ni, X. Han, L. Fan and Z. Yuan, 2015. Dissecting microbial community structure and methane-producing pathways of a full-scale anaerobic reactor digesting activated sludge from wastewater treatment by metagenomic sequencing. Microb. Cell Fact., Vol. 14.
CrossRefDirect Link - Macey, M.C., E. Curtis-Harper and K. Olsson-Francis, 2019. Draft genome sequence of Clostridium sp. Strain E02, isolated from an estuarine environment. Microbiol. Resour. Announc.
CrossRefDirect Link - Diaz, C.R., C. Seyboldt and M. Rupnik, 2018. Non-human C. difficile reservoirs and sources: Animals, food, environment. Adv. Exp. Med. Biol., 1050: 227-243.
CrossRefDirect Link - Becerra-Castro, C., G. Macedo, A.M.T. Silva, C.M. Manaia and O.C. Nunes, 2016. Proteobacteria become predominant during regrowth after water disinfection. Sci. Total Environ., 573: 313-323.
CrossRefDirect Link - Mardanov, A.V., A.V. Beletsky, Y. Nikolaev, R.Y. Kotlyarov, A. Kallistova, N.V. Pimenov, N.V. Ravin, 2017. Metagenome of the microbial community of anammox granules in a nitritation/anammox wastewater treatment system. Genome Announc.
CrossRefDirect Link - Simon, J. and P.M.H. Kroneck, 2013. Microbial sulfite respiration. Adv. Microb. Physiol., 62: 45-117.
CrossRefDirect Link - van den Brand, T.P.H., K. Roest, G.H. Chen, D. Brdjanovic and M.C.M. van Loosdrecht, 2015. Potential for beneficial application of sulfate reducing bacteria in sulfate containing domestic wastewater treatment. World J. Microbiol. Biotechnol., 31: 1675-1681.
CrossRefDirect Link - Kraft, B., M. Strous and H.E. Tegetmeyer, 2011. Microbial nitrate respiration-genes, enzymes and environmental distribution. J. Biotechnol., 155: 104-117.
CrossRefDirect Link - Russ, L., D.R. Speth, M.S.M. Jetten, H.J.M. Op den Camp and B. Kartal, 2014. Interactions between anaerobic ammonium and sulfur-oxidizing bacteria in a laboratory scale model system. Environ. Microbiol., 16: 3487-3498.
CrossRefDirect Link - Jones, Z.L., J.T. Jasper, D.L. Sedlak and J.O. Sharp, 2017. Sulfide-induced dissimilatory nitrate reduction to ammonium supports anaerobic ammonium oxidation (anammox) in an open-water unit process wetland. Appl. Environ. Microbiol., Vol. 83.
CrossRefDirect Link - Zhang, Z., H. Wang, J. Zhou, H. Li and Z. He et al., 2015. Redox potential and microbial functional gene diversity in wetland sediments under simulated warming conditions: Implications for phosphorus mobilization. Hydrobiologia, 743: 221-235.
CrossRefDirect Link - Feng, Y., R. Kumar, D.A. Ravcheev and H. Zhang, 2015. Paracoccus denitrificans possesses two BioR homologs having a role in.regulation of biotin metabolism MicrobiologyOpen, 4: 644-659.
CrossRefDirect Link - Li, A., Y. Chu, X. Wang, L. Ren and J. Yu et al., 2013. A pyrosequencing-based metagenomic study of methane-producing microbial community in solid-state biogas reactor. Biotechnol. Biofuels.
CrossRefDirect Link - Ma, Q., Q.U. Yuanyuan, X. Zhang, Z. Liu, H. Li and Z. Zhang et al., 2015. Systematic investigation and microbial community profile of indole degradation processes in two aerobic activated sludge systems. Sci. Rep.
CrossRefDirect Link - Yang, Y., K. Yu, Y. Xia, F.T.K. Lau and D.T.W. Tang et al., 2014. Metagenomic analysis of sludge from full-scale anaerobic digesters operated in municipal wastewater treatment plants. Appl. Microbiol. Biotechnol., 98: 5709-5718.
CrossRefDirect Link - Huang, Q., W.L. Du, L.L. Miao, Y. Liu and Z.P. Liu, 2018. Microbial community dynamics in an ANAMMOX reactor for piggery wastewater treatment with startup, raising nitrogen load, and stable performance. AMB Expr.
CrossRefDirect Link