
Hanan Abo El-Kassem Bosly
Department of Biology, Faculty of Science, Jazan University, Saudi Arabia
LiveDNA: 966.27899







ABSTRACT
Background and Objective: The housefly (Musca domestica L.) is a globally distributed insect in the order Diptera, family Muscidae that is well known throughout the world. The housefly is the primary cause of many epidemics in humans, domestic animals and livestock. This study was conducted in the Jazan region, Kingdom of Saudi Arabia, to study the genetic resources and obtain baseline knowledge of the fundamental molecular identification of this insect. Material and Methods: The individuals (male and female) were collected from the Abu Arish area (eastern Jazan). Total genomic DNA was extracted for polymerase chain reaction (PCR) using the specific primers Fly-F and Fly-R for the mitochondrial cytochrome oxidase subunit I gene (mtCOI). A short partial fragment of the mtCOI gene (~272 nucleotides) was successfully amplified from all samples and the amplicons were subsequently sequenced. Results: The resultant sequences encoded ~87 amino acids and represented the mtCOI gene of M. domestica according to BLAST analysis. Pairwise nucleotide sequence analysis of the partial mtCOI gene revealed its closest identity was with Singapore and Thailand isolates (99%). However, we observed no genetic variability within our species from Jazan. Phylogenetic dendrograms grouped all species together with the M. domestica species reported from Singapore and Thailand. This study provides the first mitochondrial sequence analysis of the mtCOI gene of the housefly M. domestica for insect identification in Jazan region. Conclusion: This study revealed that a short fragment of the mtCOI gene could be successfully used for the identification of the housefly.
PDF Abstract XML References Citation
Copyright: © 2020. This is an open access article distributed under the terms of the creative commons attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
How to cite this article
Hanan Abo El-Kassem Bosly, 2020. Molecular Identification of Musca domestica L. from Jazan (KSA) Based on Partial Mitochondrial Cytochrome Oxidase Gene Sequencing. Journal of Entomology, 17: 6-13.
DOI: 10.3923/je.2020.6.13
URL: https://scialert.net/abstract/?doi=je.2020.6.13
DOI: 10.3923/je.2020.6.13
URL: https://scialert.net/abstract/?doi=je.2020.6.13
INTRODUCTION
The house fly Musca domestica Linnaeus (Diptera) is one of the most studied species globally distributed and transmitting pathogenic agents in humans1,2. The composition of the genetic information contained in mitochondrial DNA has been extensively researched and can characterize a population, phylogenetic and make it possible to reconstruct evolutionary history3,4. The exact origin of the housefly remains not well investigated, however, a few studies report the Middle East as a likely origin of this species5. In Saudi Arabia, the prevalence of dipterous flies was previously surveyed for general identification6 and for forensic importance7. Based on morphological keys, previously identified 12 species of houseflies belonging to 8 genera and 7 families6. In Jazan region of Saudi Arabia no previous studies performed for identification using mitochondrial sequence analysis of the mtCOI gene of the housefly M. domestica. Meanwhile, DNA-based methods are widely used for species determination at any life cycle stage of an insect with a variety of live, dead and preserved samples8. The use of mitochondrial DNA (mtDNA) is advantageous compared with genomic DNA because of the high mutation rates and multiple copies in the cell9,10. The structural variation and differentiation in the population genetics of an organism can be evaluated by studying the changes in mtDNA5. mtDNA is maternally regulated and non-recombinant in nature and therefore has been widely utilized as a steady molecular marker to collect comparative information with other species11. The mtDNA encoding the cytochrome oxidase I (COI) gene is particularly helpful to study population genetics and evolutionary relationships of species12,13. Moreover, COI gene sequencing has been successfully employed to identify several fly species with forensic importance in different parts of the world7,8,14.
The dipteran housefly (Musca domestica L.) in the family Muscidae is morphologically and genetically a diverse, global cosmopolitan insect of human and domestic animals with medical and veterinary importance15. Houseflies serve as vectors for transmitting devastating bacterial and viral pathogens in humans and animals16. The housefly is a year long actively colonizing and global insect found mostly in tropical and temperate environments. However, the fly adapts to its local environmental conditions depending on the surrounding biogeographical climate, breeding niche and the use of insecticides17. The genetic differentiation of geographically distant housefly populations can be affected by environmental differences. The genetic diversity of housefly populations is determined by a variety of methods. Many genetic markers such as allozymes18, microsatellites19 and mtDNA7,15 are commonly used to evaluate genetic diversity. The present work was designed to establish the molecular characterization of M. domestica through analysis of a partial sequence of the mtCOI gene in the Jazan region, Kingdom of Saudi Arabia.
MATERIALS AND METHODS
Insect sample collection: The present study was conducted from October, 2018 to April, 2019. The representative samples of the housefly M. domestica (males and females) were collected from Abu Arish, Jazan region, KSA, at 42° 49 57" E, 16° 58 8.04" N. The samples were collected and preserved in 70% ethanol until further analysis as previously described by Bosly6.
Synthesis of oligonucleotides: The oligonucleotide primers used as previously described by Liu et al.20 as follow: Specific primers: Fly-F (5 -CAGATCGAAATTTAAATAC-TTC-3 ) and Fly-R (5 -GTATCAACATCTATTCCTAC-3 ).
Extraction and quantification of Musca domestica total genomic DNA: All specimens collected from Jazan region, Kingdom of Saudi Arabia. The thoracic muscles of each insect were used as the source of DNA to avoid potential DNA contamination from intestinal parasite proteins and gut eggs of parasites, whereas the residues of each sample were stored in an eppendorf tube (1.5 mL) at -20°C for identity verification and sample buildup. The total genomic DNA was extracted using 5 individuals from each sex. Total cellular DNA was extracted from previously collected flies using the CTAB method21. The total genomic DNA was successively quantified using a NanoDrop™ 2000/2000c Spectrophotometer and the purity of DNA samples was confirmed by the Amax/Amin ratio. The quality of the DNA was also evaluated by agarose gel electrophoresis. DNA samples were used for polymerase chain reaction (PCR reaction).
PCR amplification and electrophoresis analysis: An ~272 bp fragment of the mtCOI coding sequence was amplified using the Fly-F and Fly-R primers. PCR was performed in a 25 μL reaction volume containing 2 μL (10-20 ng) of template DNA, 4 μL of dNTP (1 mmol mL–1), 1.0 U of Taq polymerase (Promega, Madison, WI, USA), 2.5 μL of 10X buffer (MgCl2 1.5 mmol L–1) and 0.25-2.5 μL of each amplification primer Fly-F and Fly-R (10 μmol). The amplification was conducted using a GeneAmp PCR system 9700 thermal cycler (Applied Biosystems, USA) and 0.2 mL MicroAmp® Reaction PCR tubes. PCR was performed using the following protocol: denaturation at 94°C for 4 min, followed by 35 cycles of denaturation at 94°C for 40 sec, annealing at 48°C for 1 min and extension at 72°C for 1 min. A final extension cycle at 72°C for 10 min was conducted to ensure flush ends on the DNA molecules. The resultant PCR amplicons of the mtCOI regions were resolved by electrophoresis onto 2% agarose gel in 0.5X TBE buffer (40 mmol Tris Base, 1 mmol boric acid, 2 mmol Na2 EDTA) with ethidium bromide (10 mg mL–1). To check for the correct size of both PCR products, 6 mL of DNA sample was mixed with 1.5 mL of 6X gel loading buffer (0.25%) bromophenol blue, 0.25% xylene cyanol FF and 30% glycerol. The sizes were compared with a 100 bp gene ruler (Fermentas, Hanover, MD). DNA was visualized on a UV transilluminator (λ = 254 nm) and photographed with the InGenius LHR Gel Imaging System (Syngene, UK).
Automated DNA sequencing: After confirmation, the amplicons were purified from the agarose gel using a gel extraction kit (Qiagen QIAquick Gel, Valencia, CA) according to the manufacturer’s protocol. The purified DNA was sent directly for sequencing using the Fly-F primer in a single direction with an ABI PRISM Big Dye Terminator Cycle Sequencing Ready Reaction Kit for an ABI PRISM 3730 (Applied Biosystems, USA) with BigDye terminator v3.1 as the sequencing agent.
Nucleotide comparison and construction of phylogenetic tree: The obtained sequences were initially blasted using BLASTn in the NCBI GenBank database. The top hits were retrieved and aligned together with the candidate sequences in DNAMAN software using the clustalW algorithm to calculate the pairwise nucleotide (nt) sequence identities. The evolutionary relationships were determined by constructing a phylogenetic dendrogram using a neighbor-joining algorithm and one thousand bootstrap iterations. The evolutionary distances were computed and expressed with number of base-substitutions/site with the pairwise deletion option selected. The individual nodes of each branch were supported at 60% using a bootstrap test with 1000 replicates.
RESULTS
PCR amplification and automated partial nucleotide sequence of mt (COI) gene: The yield of total genomic DNA was determined as 120 μg/0.02 g of tissue and the purity of DNA samples was indicated by the Amax/Amin ratio of 1.7.
A partial sequence of the mtCOI gene was successfully amplified using the 2 primers Fly-F and Fly-R. An ~270 bp amplicon from each sample was amplified via PCR amplification (Fig. 1a, b). After confirmation with gel electrophoresis, each amplicon from the ten housefly samples was subsequently sequenced.
The resultant nucleotide (nt) sequences were initially analyzed using the BLASTn tool available in the NCBI GenBank database (https://www.ncbi.nlm.nih.gov). All the obtained sequences were deposited in the NCBI GenBank database and an accession number was obtained for each entry (Table 1).
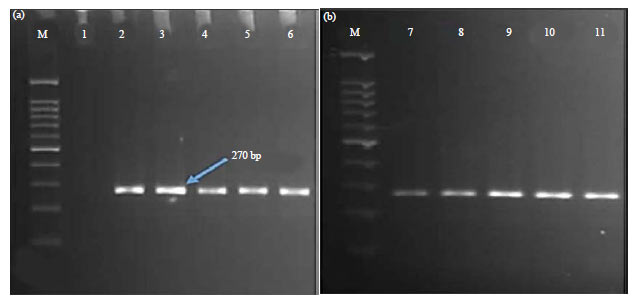 | |
| Fig. 1(a-b): | Agarose gel electrophoresis using 2% agarose gel showing the PCR products of mtCOI gene using set of primers (a) Fly-F and (b) Fly-R |
M: 100 bp Marker, Lane 1: Negative control, Lanes 2-6: Amplified PCR products from adult male houseflies, Lanes 7-11: Amplified PCR products from adult female houseflies | |
 | |
| Fig. 2: | Molecular analysis of partial mtCOI gene amplified and sequenced from housefly (M. domestica) in Jazan region, Kingdom of Saudi Arabia |
Amino acids sequence were alignments for ten sequences of mtCOI gene of M. domestica using DNAMAN 8 | |
| Table 1: | Pairwise percent (%) nucleotide and amino acid sequence identities of the partial mtCOI gene from 10 M. domestica samples with the selected sequences retrieved from NCBI GenBank database using DNAMAN tool |
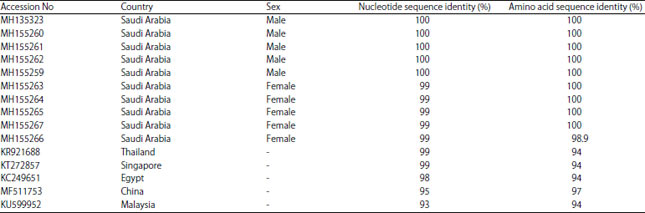 | |
The nucleotide sequences of the selected mtCOI sequences were subsequently retrieved and used to perform pairwise nt sequence comparison using the DNAMAN program (Fig. 2). The nucleotide sequence comparison revealed that the 10 isolates of the mtCOI gene sequence were 99-100% identical to one another, whereas they shared their highest nt sequence identities at 99% with the M. domestica isolates reported from Singapore (KR921688) and Thailand (KT272857) (Table 1). However, they also shared nt sequence identities at 93-98% with those reported from China (MF511753), Egypt (KC249651) and Malaysia (KU599952). Similarly, these sequences shared their highest amino acid sequence identity at 97% with those reported from China, whereas with all other isolates, they shared 94% amino acid sequence identity (Table 1). The evolutionary relationships of the M. domestica mtCOI gene species in this study were also determined using phylogenetic dendrograms of nt sequences (Fig. 3) and amino acid sequences (Fig. 3a). The phylogenetic dendrograms grouped all ten M. domestica species into a well-supported clade (99% bootstrap) with other M. domestica species reported from Thailand and Singapore (Fig. 3b). These sequences displayed identities of 93-98% with those reported from China (MF511753), Egypt (KC249651) and Malaysia (KU599952). The phylogenetic tree also identified this relationship among these species.
DISCUSSION
Characterization of the housefly genome supplies effective consequences for authorizing assignment on innovative techniques of insect surveillance, genetic engineering of repellent resistance mechanisms, genetic acclimation to altitude pathogen conception and for reconnoitering the basic biology of this important pest. Understanding the genome sequence could lead to disarrangement of the remarkable autosome-based sex deduction system, sterile male release and confounding signals for mate recognition, which offer important alternative control measures for houseflies22,23.
 | |
| Fig. 3(a-b): | Phylogenetic dendrograms of the 10-mtCOI sequences of M. domestica constructed to infer the evolutionary relationships using (a) nt sequences and (b) Amino acid sequences |
Isolates used in this study boxed in a rectangular, the nodes on each branch supported by the respective percent bootstrap values (numeric on each node) | |
The polymerase chain reaction (PCR) is promising and can be considered the most sensitive and reliable technique. Therefore, a fundamental step in the (PCR) technique remains the preparation of housefly genomic DNA. Thus, for a successful amplification process, first, fresh or dried thorax tissues were extracted. The quality of DNA isolated during this study depended on the source of the tissue used as the starting material and on the efficiency of the extraction method as previously revealed by Skevington and Yeates21. The difficulty in extracting insect DNAs is the existence of biomolecule complex (chitin, complex proteins and peptides) in the exoskeleton. This compounds may reduce the efficiency of buffers and proteinase enzymes during extraction24,25. Based on the concentration and purity of the extracted DNA, the present study demonstrated that C-TAB extraction method, which is used to extract house fly DNA, has effective in extracting total DNA from thoracic tissues. The yield of total DNA was 120 μg/0.02 g of tissue. The purity of the DNA was indicated by A260/A280 ratios. In addition to absorbance ratios, DNA quality was evaluated by agarose gel electrophoresis, which gave the characteristic 2:1 ratio of 28S-18S RNA, indicating no significant degradation. These results are consistent with prior studies by Takada et al.26 and Guo et al.27 which reported that the DNA extraction protocol was a prerequisite for successful identification of housefly genes from their respective tissues using a PCR assay.
The present study results demonstrated the successful identification of M. domestica by amplification of 270 bp of the unique mtCOI gene, indicating the accuracy of M. domestica identification in PCR assays using DNA primers for the mtCOI region. The results are consistent with those of previous studies that concluded that sequence analysis of the mitochondrial part coding the cytochrome oxidase I (COI) gene is predominately profitable in population genetics and evolutionary studies because of the relatively high level of divergence. Such an analysis is effective for restoration of evolutionary patterns, such as determination of the origin of species, the routes of invasion and historical demography. Mitochondrial DNA can be used as a proper molecular marker because of the plain manner of inheritance, relatively high rate of nucleotide substitutions and the accessibility of comparative data with other species11,17,19,28. The morphological identification keys for adult Muscidae include thorax, leg chaetotaxy, wing venation and from carrion insect29,30. However, identification based on morphological characterization is a difficult and time-consuming technique, which may cause erroneous identification for non-experts. In a previous study, dipterous flies from Jazan region in the Kingdom of Saudi Arabia, ~5000 individual flies belonged to 12 different species divided into 8 genera and 7 families have been surveyed6. Also, some other studies also characterize the housefly population based on morphological keys in Saudi Arabia31,32. The development in DNA barcoding technique has opened up new horizons for correct and trustworthy species identification33. Moreover, this technique has not been extensively applied to study the family Muscidae and only a few studies are available that employed nt sequencing data to construct a phylogenetic analysis17,34-36. The Kingdom of Saudi Arabia is an important country in the Middle East region, however, the application of molecular techniques to the genetic variation in houseflies is rarely applied. In a previous investigation by Mashaly et al.7, the abundance and prevalence of forensically important flies were studied using a DNA-bar coding technique in Saudi Arabia. In that study, a large fragment (~710 bp) of the "Folmer Region" of the mtCOI gene was used. The present study analysis clearly showed that a shorter fragment of the mtCOI gene (~270 bp) could be successfully used to identify housefly species in Jazan region, KSA.
CONCLUSION
The study inferred from the analysis that the houseflies in the Jazan region were most closely related to the Singapore and Thailand haplotypes. The study recorded low genetic variation among the houseflies in the Jazan region of Saudi Arabia. However, the study necessitates the use of other genetic markers to widen our understanding about the genetic variation of houseflies in this region. Furthermore, the study confirmed the importance of bar coding in effective identification of insect species. Moreover, the morphological and molecular identification of insects must be considered of equal importance for any taxonomic study.
SIGNIFICANCE STATEMENT
This study discovers that the houseflies in the Jazan region are most closely related to that of Singapore and Thailand haplotypes. The study confirms the importance of bar coding in effective identification of insect species. In addition to the equal importance of the morphological and molecular identification of insects for any taxonomic study.
ACKNOWLEDGMENT
The author acknowledge Associate Professor Adel A Rezk Department of biotechnology, King Faisal University, Al-Ahsa, Al Hofuf, Saudi Arabia, for help in DNA sequencing.
REFERENCES
- Kassiri, H., K. Akbarzadeh and A. Ghaderi, 2012. Isolation of pathogenic bacteria on the house fly, Musca domestica L. (Diptera: Muscidae), body surface in Ahwaz hospitals, Southwestern Iran. Asian Pac. J. Trop. Biomed., 12: S1116-S1119.
CrossRefDirect Link - Nazari, M., T. Mehrabi, S.M. Hosseini and M.Y. Alikhani, 2017. Bacterial contamination of adult house flies (Musca domestica) and sensitivity of these bacteria to various antibiotics, captured from Hamadan City, Iran. J. Clin. Diagn. Res., 11: DC04-DC07.
CrossRefPubMedDirect Link - Hebert, P.D., S. Ratnasingham and J.R. de Waard, 2003. Barcoding animal life: Cytochrome c oxidase subunit 1 divergences among closely related species. Proc. R. Soc. London B: Biol. Sci., 270: S96-S99.
CrossRefPubMedDirect Link - Bernasconi, M.V., C. Valsangiacomo, J.C. Piffaretti and P.I. Ward, 2000. Phylogenetic relationships among Muscoidea (Diptera: Calyptratae) based on mitochondrial DNA sequences. Insect Mol. Biol., 9: 67-74.
CrossRefDirect Link - Marquez, J.G. and E.S. Krafsur, 2002. Gene flow among geographically diverse housefly populations (Musca domestica L.): A worldwide survey of mitochondrial diversity. J. Heredity, 93: 254-259.
CrossRefDirect Link - Bosly, H.A.M., 2010. Prevalence of dipterous flies with veterinary importance in selected sheep's farms and slaughter houses in Jazan, Saudi Arabia. Egypt. Acad. J. Biol. Sci., 3: 63-73.
CrossRefDirect Link - Mashaly, A., R. Alajmi, A.E.Z. Mustafa, A. Rady and H. Alkhedir, 2017. Species abundance and identification of forensically important flies of Saudi Arabia by DNA barcoding. J. Med. Entomol., 54: 837-843.
CrossRefDirect Link - Aly, S.M. and J. Wen, 2013. Applicability of partial characterization of cytochrome oxidase I in identification of forensically important flies (Diptera) from China and Egypt. Parasitol. Res., 112: 2667-2674.
CrossRefDirect Link - Waugh, J., 2007. DNA barcoding in animal species: Progress, potential and pitfalls. BioEssays, 29: 188-197.
CrossRefDirect Link - Boehme, P., J. Amendt and R. Zehner, 2012. The use of COI barcodes for molecular identification of forensically important fly species in Germany. Parasitol. Res., 110: 2325-2332.
CrossRefDirect Link - Dogac, E., I. Kandemir and V. Taskin, 2013. The genetic polymorphisms and colonization process of olive fly populations in Turkey. PLoS ONE, Vol. 8.
CrossRefDirect Link - Malacrida, A.R., L.M. Gomulski, M. Bonizzoni, S. Bertin, G. Gasperi and C.R. Guglielmino, 2007. Globalization and fruitfly invasion and expansion: The medfly paradigm. Genetica, Vol. 131.
CrossRefDirect Link - Aly, S.M. and S.M. Mahmoud, 2016. COII “long fragment” reliability in characterisation and classification of forensically important flies. Arch. Forensic Med. Criminol., 66: 95-105.
CrossRefPubMedDirect Link - Kavitha, R., W.A. Nazni, T.C. Tan, H.L. Lee and M.S. Azirun, 2013. Review of forensically important entomological specimens collected from human cadavers in Malaysia (2005-2010). J. Forensic Legal Med., 20: 480-482.
CrossRefDirect Link - Cummings, M.A. and E.S. Krafsur, 2005. Spatial diversity in mitochondrial cytochrome c oxidase in house flies. Med. Vet. Entomol., 19: 53-59.
CrossRefDirect Link - Ugbogu, O.C., N.C. Nwachukwu and U.N. Ogbuagu, 2006. Isolation of Salmonella and Shigella species from house flies (Musca domestica L.) in Uturu, Nigeria. Afr. J. Biotechnol., 5: 1090-1091.
Direct Link - Dogac, E., 2016. Mitochondrial genetic variations in natural house fly (Musca domestica L.) populations from the Western and Southern parts of Turkey. Mitochondrial DNA Part A: DNA Mapp. Sequencing Anal., 27: 3802-3807.
CrossRefDirect Link - Scott, J.G., W.C. Warren, L.W. Beukeboom, D. Bopp and A.G. Clark et al., 2014. Genome of the house fly, Musca domestica L., a global vector of diseases with adaptations to a septic environment. Genome Biol., Vol. 15.
CrossRefDirect Link - Krafsur, E.S., M.A. Cummings, M.A. Endsley, J.G. Marquez and J.D. Nason, 2005. Geographic differentiation in the house fly estimated by microsatellite and mitochondrial variation. J. Heredity, 96: 502-512.
CrossRefDirect Link - Liu, Q., J. Cai, Y. Guo, X. Wang and Y. Gu et al., 2011. Identification of forensically significant calliphorids based on mitochondrial DNA Cytochrome Oxidase I (COI) gene in China. Forensic Sci. Int., 207: e64-e65.
CrossRefPubMedDirect Link - Skevington, J.H. and D.K. Yeates, 2000. Phylogeny of the Syrphoidea (Diptera) inferred from mtDNA sequences and morphology with particular reference to classification of the Pipunculidae (Diptera). Mol. Phylogenet. Evol., 16: 212-224.
CrossRefDirect Link - Ying, B.W., T.T. Liu, H. Fan, D. Wei and F.Q. Wen et al., 2007. The application of mitochondrial DNA cytochrome oxidase II gene for the identification of forensically important blowflies in Western China. Am. J. Forensic Med. Pathol., 28: 308-313.
CrossRefDirect Link - Meiklejohn, K.A., J.F. Wallman and M. Dowton, 2011. DNA-based identification of forensically important Australian Sarcophagidae (Diptera). Int. J. Legal Med., 125: 27-32.
CrossRefDirect Link - Manuahe, C., M.Y. Semuel and V.I.Y. Roring, 2016. Optimization of DNA extraction and the position of mosquito species from Southeast Minahasa in North Sulawesi using NADH dehydrogenase gene and cytochrome oxidase sub unit 1 gene. J. Entomol. Zool. Stud., 4: 498-508.
Direct Link - Timah, S. and M.Y. Semuel, 2017. Morphometry and phylogeny reconstruction Aedes sp. based DNA mitochondrial cytochrome oxidase gene sub unit 1 (CO1) in North Sulawesi. Int. J. Mosq. Res., 4: 98-106.
Direct Link - Takada, Y., T. Hiroyoshi and M. Hirano, 1988. Linkage group analysis of permethrin resistance in the Miyakonojo colony of the housefly, Musca domestica L. (Diptera: Muscidae). Applied Entomol. Zool., 23: 122-126.
CrossRefDirect Link - Guo, Y., J. Cai, Y. Chang, X. Li and Q. Liu et al., 2011. Identification of forensically important sarcophagid flies (Diptera: Sarcophagidae) in China, based on COI and 16S rDNA gene sequences. J. Forensic Sci., 56: 1534-1540.
CrossRefDirect Link - Xiong, F., Y. Guo, B. Luo, L. Zhang, J. Cai, X. Li and Z. Yang, 2012. Identification of the forensically important flies (Diptera: Muscidae) based on cytochrome oxidase subunit I (COI) gene in China. Afr. J. Biotechnol., 11: 10912-10918.
Direct Link - Grzywacz, A., M.J.R. Hall, T. Pape and K. Szpila, 2017. Muscidae (Diptera) of forensic importance-an identification key to third instar larvae of the western Palaearctic region and a catalogue of the muscid carrion community. Int. J. Legal Med., 131: 855-866.
CrossRefDirect Link - Grzywacz, A., J. Ogiela and A. Tofilski, 2017. Identification of Muscidae (Diptera) of medico-legal importance by means of wing measurements. Parasitol. Res., 116: 1495-1504.
CrossRefDirect Link - Al-Ghamdi, K.M., M. Alikhan, J.A. Mahyoub, N.A. Alanazi, A.R. Al-Najada, M.I. Nassar and B.Z. Alfarhan, 2015. Characterization of forensically important necrophagus flies (Diptera) of Jeddah, Saudi Arabia. Adv. Environ. Biol., 9: 58-71.
Direct Link - Mashaly, A.M., 2016. Entomofaunal succession patterns on burnt and unburnt rabbit carrion. J. Med. Entomol., 53: 296-303.
CrossRefDirect Link - Marquez, J.G., M.A. Cummings and E.S. Krafsur, 2007. Phylogeography of stable fly (Diptera: Muscidae) estimated by diversity at ribosomal 16S and Cytochrome Oxidase I mitochondrial genes. J. Med. Entomol., 44: 998-1008.
CrossRefDirect Link - Kutty, S.N., T. Pape, A. Pont, B.M. Wiegmann and R. Meier, 2008. The Muscoidea (Diptera: Calyptratae) are paraphyletic: Evidence from four mitochondrial and four nuclear genes. Mol. Phylogenet. Evol., 49: 639-652.
CrossRefDirect Link - Guo, Y.D., J.F. Cai, F.M. Meng, Y.F. Chang and Y. Gu et al., 2012. Identification of forensically important flesh flies based on a shorter fragment of the cytochrome oxidase subunit I gene in China. Med. Vet. Entomol., 26: 307-313.
CrossRefDirect Link - Bosly, A.H., 2018. Molecular identification studies on Oestrus ovis L.(Diptera: Oestridae) larvae infested sheep in Jazan region, Saudi Arabia. Indian J. Anim. Res., 52: 105-110.
CrossRefDirect Link