
Murali R Kuracha
Department of Zoology, Osmania University College for Women, Koti, Hyderabad, Andhra Pradesh, India
Bhargavi Rayavarapu
Department of Zoology, Osmania University College for Women, Koti, Hyderabad, Andhra Pradesh, India
Venkata Rama Satya Kumar Duvvuri
Department of Epidemiology and Preventive Medicine, University of Maryland, Baltimore, Maryland, USA
Poduri Nagaraja Rao
Department of Zoology, Osmania University College for Women, Koti, Hyderabad, Andhra Pradesh, India
Journal of Entomology
Year: 2006 | Volume: 3 | Issue: 3 | Page No.: 222-230







ABSTRACT
A comparative study of common core secondary structure in the ribosomal Internal Transcriber 2 (ITS2) of 8 Lepidopteran species selected from different geographical locations was carried out. Among the selected insects some are serious pests of agricultural crops. Ex: Helicoverpa punctigera. Multiple sequence alignment and secondary structural analysis of ITS2 was performed to elucidate the phylogenetic relationship. These studies indicated a phylogenetic relationship among the selected insects belonging to different geographical locations. Several common features of secondary structure are shared among these species, with some of them supported by compensatory changes, suggesting the significant role by ITS2 as an RNA domain during ribosome biogenesis.
PDF Abstract XML References
How to cite this article
Murali R Kuracha, Bhargavi Rayavarapu, Venkata Rama Satya Kumar Duvvuri and Poduri Nagaraja Rao, 2006. Comparison of Secondary Structure of the Ribosomal Internal Transcribed Spacer 2 of Eight Lepidopteran species from Diverse Geographical Locations. Journal of Entomology, 3: 222-230.
DOI: 10.3923/je.2006.222.230
URL: https://scialert.net/abstract/?doi=je.2006.222.230
DOI: 10.3923/je.2006.222.230
URL: https://scialert.net/abstract/?doi=je.2006.222.230
INTRODUCTION
Lepidopteran insects are basically phytophagous in nature. The larvae of these lepidopteran insects are voracious. Hence, they have become major pests of various crops (Metcalf and Flint, 1962; Nair, 1986; David, 1991; Srivastava, 2000). Intra-specific conservation and variations have been reported using ITS2 region collected from diverse geographic locations that are used for phylogenetic studies (Van der Sande et al., 1992; Michot et al., 1984; Shinohara et al., 1999; Crabtree et al., 1995). The internal transcribed spacers (ITS) are located between the repeating array of nuclear 18S, 5.8S and 28S ribosomal RNA genes, a locus that has 100-200 copies per genome. The ITS spacers are versatile genetic markers and have been used for phylogenetic analysis, evaluation of the evolutionary process, as well as for determination of taxonomic identities (Atanas and Nazar, 1999). The lepidopteran insects selected for the present study were: Helicoverpa punctigera, Luehdorfia longicaudata, Luehdorfia chinensis, Luehdorfia puziloi puziloi, Luehdorfia puziloi yessoensis, Heliconius charithonia, Heliconius hecale and Heliconius melpomene.
Mature rRNAs are produced by the processing of a large precursor from which different transcribed spacer regions are sequentially removed through an elaborate pathway of cleavage steps (Perry, 1976). In eukaryotes, transcribed spacer regions may represent a very substantial fraction of the length of the primary transcript. Although these transcribed spacer regions are obvious candidates for important roles in the control of ribosome biogenesis, elucidation of their biological function and of the molecular mechanisms involved in their accurate excision still remain a major challenge. Recent functional analyses performed on yeast ribosomal RNA genes clearly show that the structural integrity of the transcribed spacer regions is an essential prerequisite for correct processing of mature rRNA and biogenesis of active ribosomal subunits (Michot et al., 1984; Musters et al., 1990). The derivation of reliable secondary structure models for each transcribed spacer region would undoubtedly represent a major step towards a detailed understanding of their biological role. The comparative sequence analysis provides the most powerful tool for identifying the biologically relevant folding pattern of an RNA molecule, i.e., its native structure within the cellular context (Michot et al., 1984). However, an essential prerequisite for its effective utilization is the availability of a collection of sequences exhibiting a substantial number of nucleotide differences while remaining similar enough for unequivocal sequence alignments. Due to the high rate of sequence variation of transcribed spacers, this may exhibit dramatic size variation and extensive sequence divergence even among moderately distant species (Michot et al., 1983; Furlong and Maden, 1983). Nevertheless, the presence of phylogenetically conserved secondary structure elements in the 5’ externally transcribed spacer was recently revealed by the comparative analysis of a limited set of vertebrate sequences (Michot and Bachellerie, 1991).
The present study is focused on internal transcribed spacer, ITS2, which interrupts the eukaryotic large subunit rRNA molecule and has no prokaryotic equivalent (Michot et al., 1984). The sequence and secondary structures of ITS2 of selected geographically variant Lepidopteran insect species were comprehensively investigated. Such case studies are relevant in broader phylogenetic contexts and for analyzing the function in ribosome biogenesis. Since the secondary structures of ITS region are more conserved than the nucleotide sequences their analysis helps in understanding molecular evolution and increases the number of structural characters. Thus the structure models developed in this study can be used for future phylogenetic analysis.
MATERIALS AND METHODS
Data Set
ITS 2 sequences of eight Lepidopteran species belonging to diverse geographical locations (Costa Rica, China, Russia, America and Japan) that are deposited in GenBank were investigated. The accession numbers of Helicoverpa punctigera, Luehdorfia longicaudata, Luehdorfia chinensis, Luehdorfia puziloi puziloi, Luehdorfia puziloi yessoensis, Heliconius charithonia, Heliconius hecale and Heliconius melpomene are: AF047759, AB071926, AB071925, AB071923, AB071911, AF453773, AF453768 and AF453767, respectively.
Sequence Alignments
Multiple sequence alignments were performed using CLUSTALW with a gap opening penalty of 15 and gap extension penalty of 6.66.
Secondary Structure Prediction
The RNA secondary structures for ITS2 were predicted using RNADRAW (Christoffersen et al., 1994). RNADRAW predicts RNA structures by identifying suboptimal structures using the free energy optimization methodology at a default temperature of 37°C. In the current study, ITS2 and 5.8S regions (the first 170 nucleotides) were used for RNA structure prediction. The minimum energy structure prediction algorithm in RNADRAW was ported from the RNAFOLD program included in the Vienna RNA package (Hofacker et al., 1994). The dynamic programming algorithm employed in RNADRAW was based on the work of Zuker and Stiegler (1981) and uses energy parameters taken from Freier et al. (1986) and Jaeger et al. (1989).
RNA Fold
The Sribo program in Sfold (Statistical Folding and Rational Design of Nucleic Acids) was used to predict the probable target accessibility sites (loops) for trans-cleaving ribozymes in ITS2 (Ding et al., 2004). The prediction of accessibility is based on a statistical sample of the Boltzmann ensemble for secondary structures. Here, we assessed the likelihood of unpaired sites for potential ribozyme target. Each mRNA exists as a population of different structures. Hence, stochastic approach to the evaluation of accessible sites was found appropriate (Christoffersen et al., 1994). The probability profiling approach by Ding and Lawrence (2001) reveals target sites that are commonly accessible for a large number of statistically representative structures in the target RNA. This novel approach bypasses the long-standing difficulty in accessibility evaluation due to limited representation of probable structures due to high statistical confidence in predictions. The probability profile for individual bases (W = 1) is produced for the region that includes a triplet and two flanking sequences of 15 bases each in every site of the selected cleavage triplet (e.g., GUC).
Phylogenetic Analysis
The phylogenetic Genebee service was used for phylogenetic tree construction (Brodsky et al., 1992).
RESULTS
Sequence Analysis
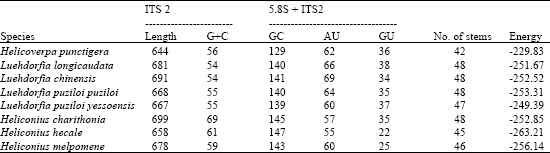
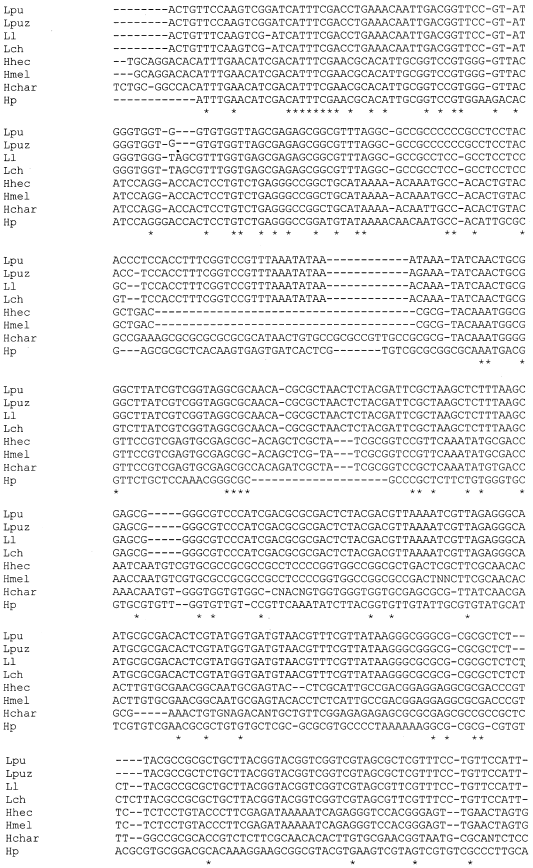
The length of ITS2 elements of eight selected lepidopteran species ranged in size between 644 and 699 bp. eight dispersed but unambiguously conserved sequence segments encompassing about a third of the ITS2 length have been identified. They were interspersed with variable regions and gaps where size variations accumulate. The characteristics of the sequences for each species are shown in Table 1. The length variations were observed with maximum length being 699 bp and minimum of 644 bp for Heliconius charithonia and Helicoverpa punctigera respectively. The G+C contents for the two regions of rDNA (5.8S and ITS2) of all species ranged from 54 to 69%. For ITS2 regions the sequence identities ranges, with maximum 99% similarity between Luehdorfia longicaudata and Luehdorfia chinensis; 95% between Luehdorfia longicaudata and Luehdorfia puziloi puziloi whereas the minimum being the 2% between Luehdorfia puziloi puziloi and Heliconius hecale. Alignment of ITS2 region is shown in the Fig. 1a. Simple tandem repeats were present at various locations along the ITS2. The sequence similarity is more towards the 5’ end and with dispersed conserved ness in the middle than towards the 3’ end.
Secondary Structure
Secondary structural features of ITS2 regions were given in the Table 1 and Fig. 2. The secondary structures of the mentioned Lepidopteran species were classified into three groups based on the analysis of conserved stems and loops. Class I includes L. longicaudata, L. chinensis, L. puziloi yessoensis, L. puziloi puziloi that show overall similarity in the ITS2 rDNA folding where L. longicaudata and L. chinensis have identical secondary structures.
| Table 1: | Lengths (in m); G+C content (in %); GC, AU, GU base pairs (in numbers); No: stems and energies (in kcal) of secondary structures of the second internal transcribed spacer (ITS-2) rDNA sequences of the Lepidopteran species |
 | |
 |
 | |
| Fig. 1a: | Alignment between Lepidopteran species ITS2 regions |
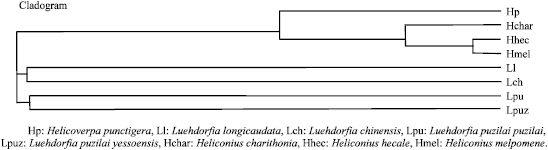 | |
| Fig. 1b: | Phyloginetic tree of B selected lepidopteran species |
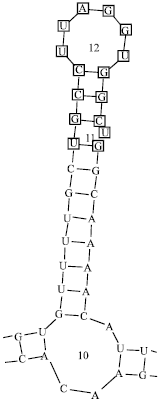 | |
| Fig. 2: | A secondary structural fold found in common to all 8 Lepidateran species. Identical bases are marked |
Secondary structures of remaining species are highly variant. Two common motifs, having sequences UGUCG and CUUCGGUG respectively were conserved in all classes. Apart from the common conserved motifs shared among the species that are categorized into different classes, variable regions also do exist. The observed similarities at the secondary structural level are further reflected at energy level.
DISCUSSION
The selected 8 Lepidopteran insect species occur worldwide and they are pests of various crops. In the present investigation, the ITS2 sequences reflected the trend observed in the phylogeny. The more distantly related the less was the convergence at the ITS2 level (Fig. 1a and b). However accumulated substitutions in the ITS sequence leading to length variation also had a profound effect on the conservedness among the structures. The length variation observed was may be due to insertions effected by many factors including genetic drift, the relative number and size of repeats, rates of unequal crossover, gene conversion, immigration and the number of the loci (Levinson and Gutman, 1987). But still high level of sequence conservation was found between some species like Luehdorfia longicaudata and Luehdorfia chinensisi, Luehdorfia longicaudata and Luehdorfia puziloi puziloi, (Severini et al., 1996) even the simple tandem repeats were found to be conserved to a large extent among them. This conservation was further reflected at secondary structural and energy level. The predicted features of ITS2 using RNADRAW are given in Table 1. The stems (double stranded paired regions) stabilize RNA secondary structures and the number of stems present in each ITS2 is given in Table 1. ITS2 RNA structures from Heliconius hecale and Heliconius melpomene the highest negative free energy (-263.21 kcal and -256.14 kcal) followed by Luehdorfia puziloi puziloi (-253.31), Heliconius charithonia (-252.85) Luehdorfia chinensis (-252.52) and then by Luehdorfia longicaudata (-251.67), Luehdorfia puziloi yessoensis (-249.39) and Helicoverpa punctigera (-229.83). Visual comparison shows that this is related to the trend in the cladogram given in Fig. 1a. This convergence at secondary structural level among few species from different geographic isolates is may be due to the evolutionary pressure on ITS2 to maintain the RNA secondary structure involved in post-transcriptional processing of rRNA (Shinohara et al., 1999). Secondary structure predictions for the ITS2 region indicate that these domains base pair to form a core region central to several stem features implying that conserved ness is more important for the proper rRNA folding pattern (Wesson et al., 1992). In contrast to the present observation with respect to Class I, Barker in his study found that (Barker, 1998) ITS 2 is unique in the 16 populations, of Rhipicephalus and Boophilus species, with considerable nucleotide variation among species and genera. However intraspecific variation of ITS2 sequences was also found in the populations. Analysis of ITS spacers of T. rangeli and T. cruzi allowed the distinction of two distinct groups, revealing a low-level similarity between them (Fernandes et al., 1999). Table 2 shows the distribution of different types of loops (hairpin, bulge, multi branched, interior and exterior) among different isolates. The segments of the ITS2 having score = 50 are further probed carefully for target site to assess the likelihood of unpaired segments. Interestingly, the observed phylogenetic trend was identified with respect to the target accessibility sites for the eight Lepidopteron isolates. The order of preference is interior loop, bulge loop, multiple branched loop, hairpin loop and exterior loops in the all the isolates.
These results suggest that the differences and conserved ness observed between ITS-2 of different species are not “neutral” and are not simple accumulated random nucleotide changes, but bear a significant functional load. In the previous study of three related mosquito genera (Aedes, Psorophora and Haemogogus), (Wesson et al., 1992) it was found that intra spacer variable regions appear to co-evolve and that ITS-2 variation is constrained to some extent by its secondary structure. Further studies on yeast (Van der Sande et al., 1992) have demonstrated that the ITS-2 is essential for the correct and efficient processing and maturation of certain ribosomal units. Furthermore, information for the efficient removal of ITS-2 from its RNA precursor is dispersed through the entire ITS-2 region and indels that effect secondary structure differentially alters rRNA processing. Critical changes in the rRNA folding pattern brought about by sequence evolution in the ITS spacer regions may thus have an important influence on the kinetics of precursor rRNA formation and ultimately on the efficient functioning of the rDNA cluster.
| Table 2: | Distribution of different types of loops [Hairpin (H), Bulge (B), Multi branched (M), Interior (I) and Exterior (E)] among different isolates of Lepidopteron species |
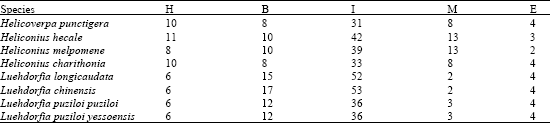 | |
CONCLUSIONS
The present study shows two contrasting aspects of ITS2 regions i.e. the general trend of variability among the species as well as the conservedness between few species. Surprisingly, the species displaying the conservedness belong to different geographical locations with diverse climatic and ecological conditions. The study implies that the ITS2 regions though have less selective pressure than the ribosomal regions but still evolve slower than the intergenic spacers, indicating that some selective pressure do exists on them, probably from the constraint to maintain the RNA secondary structure required for post-transcriptional processing and are more species specific than geographically influenced. Several common structural folds were shared among the selected lepidopteran insects for maintaining functional equivalents. Identifying the homologous regions and reconstructing their evolution increases the traits available for the phylogenetic analysis. Construction of an evolutionary tree using more isolates of Lepidopteron will provide an understanding for their functional selection. The present study indicates the phylogenetic relationship among the selected Lepidopteron species belonging to diverse geographical locations.
REFERENCES
- Atanas, I.L. and R.N. Nazar, 1999. Structural equivalence in the transcribed spacers of pre-rRNA transcripts in Schizosaccharomyces pombe. Nucleic Acids Res., 27: 3071-3078.
Direct Link - Barker, S.C., 1998. Distinguishing species and populations of rhipicephaline ticks with its 2 ribosomal RNA. J Parasitol., 84: 887-892.
PubMedDirect Link - Crabtree, M.B., H.M. Savage and B.R. Miller, 1995. Development of a species-diagnostic polymerase chain reaction assay for the identification of Culex vectors of St. Louis encephalitis virus based on interspecies sequence variation in ribosomal DNA spacers. Am. J. Trop. Med. Hyg., 53: 105-109.
Direct Link - Fernandes, O., S. Santos, A. Junqueira, A. Jansen and E. Cupolillo et al., 1999. Populational heterogeneity of Brazilian Trypanosoma cruzi isolates revealed by the mini-exon and ribosomal spacers. Mem. Inst. Oswaldo Cruz., 94: 195-197.
CrossRefDirect Link - Ding, Y. and C.E. Lawrence, 2001. Statistical prediction of single-stranded regions in RNA secondary structure and application to predicting effective antisense target sites and beyond. Nucleic Acids Res., 29: 1034-1046.
PubMedDirect Link - Freier, S.M., R. Kierzek, J.A. Jaeger, N. Sugimoto, M.H. Caruthers, T. Neilson and D.H. Turner, 1986. Improved free-energy parameters for predictions of RNA duplex stability. Proc. Nat. Acad. Sci., 83: 9373-9377.
Direct Link - Furlong, J.C. and B.E. Maden, 1983. Patterns of major divergence between the internal transcribed spacers of ribosomal DNA in Xenopus borealis and Xenopus laevis and of minimal divergence within ribosomal coding regions. EMBO J., 2: 443-448.
Direct Link - Hofacker, I.L., W. Fontana, P.F. Stadler, L.S. Bonhoeffer, M. Tacker and P. Schuster, 1994. Fast folding and comparison of RNA secondary structures. Monatsh. Chem., 125: 167-188.
CrossRefDirect Link - Jaeger, J.A., D.H. Turner and M. Zuker, 1989. Improved predictions of secondary structures for RNA. Proc. Natl. Acad. Sci. USA., 86: 7706-7710.
Direct Link - Levinson, G. and G.A. Gutman, 1987. Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol., 4: 203-221.
PubMedDirect Link - Michot, B., J.P. Bachellerie and F. Raynal, 1983. Structure of mouse rRNA precursors. Complete sequence and potential folding of the spacer regions between 18S and 28S rRNA. Nucleic Acids Res., 11: 3375-3391.
Direct Link - Michot, B. and J.P. Bachellerie, 1991. Secondary structure of the 5' external transcribed spacer of vertebrate pre-rRNA. Presence of phylogenetically conserved features. Eur. J. Biochem., 195: 601-609.
Direct Link - Michot, B., N. Hassouna and J.P. Bachellerie, 1984. Secondary structure of mouse 28S rRNA and general model for the folding of the large rRNA in eukaryotes. Nucleic Acids Res., 12: 4259-4279.
Direct Link - Musters, W., K. Boon, C.A. van der Sande, H. van Heerikhuizen and R.J. Planta, 1990. Functional analysis of transcribed spacers of yeast ribosomal DNA. EMBO J., 9: 3989-3996.
Direct Link - Wesson, D.M., C.H. Porter and F.H. Collins, 1992. Sequence and secondary structure comparisons of ITS rDNA in mosquitoes (Diptera: Culicidae). Mol. Phylogen. Evol., 1: 253-269.
Direct Link