
Hamid Ghajarieh
Department of Plant Protection, Aburayhan Compas, University of Tehran Tehran P.O. Box, Iran
Mike Bruford
Department of Ecology, School of Bioscience, Cardiff University, UK
Hassan
Department of Biology, King Khalid University, Saudi Arabia
LiveDNA: A. Dawah
Carlos
Department of Ecology, School of Bioscience, Cardiff University, UK
LiveDNA: Fernandesand Ciara S. Dodd
Journal of Entomology
Year: 2006 | Volume: 3 | Issue: 2 | Page No.: 167-179







ABSTRACT
The aim of this study was to investigate the taxonomic limits of species within four Eurytomid genera, namely Eurytoma Illiger, Tetramesa Walker, Ahtola Claridge and Sycophila (Walker). In order to further clarify the taxonomic status of the genera, including Tetramesa, Eurytoma, Ahtola and Sycophila, mitochondrial DNA sequence analysis revealed differentiation between the above Eurytomid genera. With the sequence data, the monophyletic status of three of these genera is well supported by all methods of phylogenetic reconstruction. Only Tetramesa, which is by far the most specious taxon studied here, seems polyphyletic, but even in this case, there are certain relationships within the group that are interesting from both a morphological and host preference point of view. For example, T. petiolata (Walker) and T. airae (von Schlechtendal), which are both herbivores of the grass, Deschampsia cespitosa (L.) (Beauv.) group together and two clusters contained all species that use Elymus repens (L.), i.e., T. linearis (Walker), T. hyalipenis (Walker) and T. cornuta (Walker). Another result of this analysis supports the validity of Ahtola as a separate genus, which was previously uncertain.
PDF Abstract XML References
How to cite this article
Hamid Ghajarieh, Mike Bruford, Hassan and Carlos, 2006. Mitochondrial Phylogenetics of UK Eurytomids. Journal of Entomology, 3: 167-179.
DOI: 10.3923/je.2006.167.179
URL: https://scialert.net/abstract/?doi=je.2006.167.179
DOI: 10.3923/je.2006.167.179
URL: https://scialert.net/abstract/?doi=je.2006.167.179
INTRODUCTION
Among all the chalcids, the systematics of the family Eurytomidae is particularly difficult since traditional taxonomic procedures based on morphology usually cannot identify of species group differences (Claridge, 1988). Within the family, both the classification at generic and sub-generic levels and the microtaxonomy of certain species have been a matter of disagreement among authors (Henneicke et al., 1989). Examples concern are the validity of the genus Ahtola (Claridge, 1961; Fitton et al., 1978), the limits of some genera such as Eurytoma and Tetramesa (Boucek, 1988) and the putative existence of aggregates of sibling species classified under single names (Claridge and Askew, 1960).
Acknowledging the difficulties associated with the morphological identification of Eurytomids, specialists in this research field have turned to methods of molecular systematics, but so far using only electrophoretic analysis protein (Dawah, 1988; Claridge and Dawah, 1994; Al-Barrak et al., 2004) and random amplified polymorphic DNA markers (RAPDs) (Al-Barrak et al., 2004). However, different molecular approaches, e.g., sequencing of mitochondrial DNA mtDNA have been used successfully in studies of other Chalcidoid wasps where morphological and behavioural traits are inadequate to resolve phylogenies (Gauthier et al., 2000), to detect diversity within taxa (Taylor et al., 1997), or to understand co-evolutionary processes (e.g., Agaoninae wasp pollinators of Ficus spp.) (Herre et al., 1996; Weiblen, 2001).
Mitochondrial DNA is one of the most widely used genetic markers for the study of inter-and intra-specific evolution of animals (Posada and Crandall, 1998; Simon et al., 1994; Taberlet et al., 1996) and rapid screening of arthropod mithocondrial genomes for rearrangements (Roehrdanz, 2004). Several factors have contributed to this popularity. These include its compact size (15000-17000 bp), relatively conserved gene order and number, essentially non-recombining uniparental pattern of inheritance and few insertion/deletion events (Avise et al., 1987; Harrison, 1987). Probably the most important reason for its widespread use is its rate and mode of evolution (Aquadro et al., 1984; Moritz et al., 1987). Different rates of nucleotide substitution in different gene segments and between evolutionary lineages are well-known features of mitochondrial genes and is probably due to gene-specific selection that limits divergence over evolutionary time (Lopez, 1997).
Population level studies have exploited the genome's rapid evolutionary rate, relying especially upon substitutions, which accumulate rapidly in third codon positions of coding genes (Palsbøll and Arctander, 1998). However, species and higher level relationships can be studied using the slower amino acid replacement changes (Moritz et al., 1987) and protein coding genes may be very useful for both within and between species studies (Villablanca, 1994).
The relatively fast rate of nucleotide substitution and haploid (single copy) inheritance, which reduces the effective population size of this marker and increases its sensitivity to genetic drift, make mtDNA useful for revealing phylogenetic structure between populations of species (Moritz, 1994). MtDNA phylogenies can reveal patterns of genetic differentiation among cryptic species (Moritz et al., 1993), such as may be the case in parasitic wasps.
Today, some mitochondrial genes have a universal status for certain applications and are now routinely applied in many insect molecular studies. For instance, the COI (Cytochrome Oxidase I) and COII (Cytochrome Oxidase II) genes have become the markers of choice in the study of intergeneric and interspecific relationships (Crozier et al., 1989; Liu and Beckenbach, 1992; Willis et al., 1992; Brown et al., 1994; Danforth, 1999).
In this study, during 2001-2003, the limits and phylogenetic relationships of four Eurytomid genera in the UK (Tetramesa, Eurytoma, Sycophila, Ahtola) were investigated by phylogenetic analysis of a COI-COII DNA sequence fragment from several populations from South Wales and several isolated sites in England.
MATERIALS AND METHODS
Specimen Preparation for Molecular Studies
After dissecting the stems, the larvae transferred to the gelatin capsule for rearing and then in early summer when they emerged, two-three days-old adults were collected and stored in small plastic tubes and deep-frozen at -70°C and labeled with a code number. Samples used in this study and their origins and plant hosts are listed in Table 1.
DNA Extraction
Purified DNA for the wasp species examined was obtained the QIAamp DNA Mini-kit (catalogue number 51306). All extractions followed t he manufacturer’s recommendations QIAamp DNA Mini Kit (Qiagen) protocol and specimens were homogenized in 180 μL of buffer ATL (Qiagen).
| Table 1: | Hymenoptera species used in the molecular analysis, showing collection location and host grass species. The last two species in column on left (C. BIS and C.CAP) are reference species |
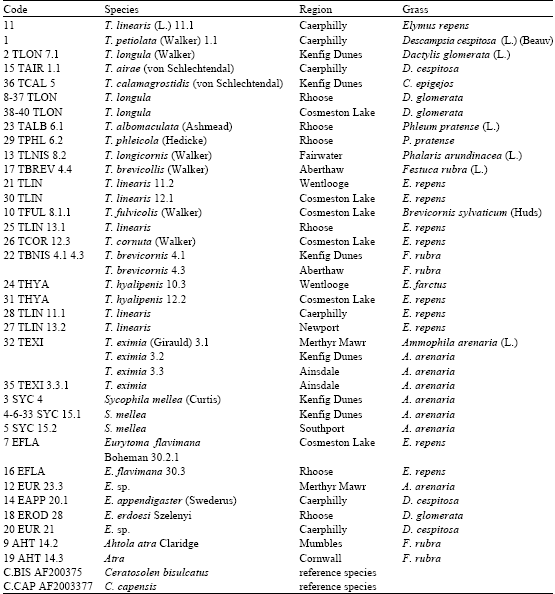 | |
| Table 2: | List of the PCR primers for amplifying the COI and COII genes. First two primers are sited in the COI, the last two in the CO II gene used |
 | |
Homogenization was performed in 1.5 mL plastic microcentrifuge tubes using a hand held pellet mixer followed by addition of 20 μL proteinase K (20 mg mL-1). Samples were mixed by vortexing and incubated in a hybridization oven containing a rocking platform at 56°C overnight. Two hundred microliter of buffer AL (Qiagen) was added to the each sample and mixed by pulse-vortexing for 15 sec followed by incubation at 70°C for 10 min. Two hundred microliter of 100% absolute ethanol was then added to each tube and mixed. The mixtures (including the precipitate), but no insect remains, were carefully applied to the QIAamp spin columns without wetting the rim. Thereafter, centrifugation was performed at 8000 rpm for 1 min. The spin columns were placed in clean collection tubes. Without wetting the rim, 500 μL of buffer AW1 (Qiagen) was added to the QIAamp spin columns and centrifuged at 8000 rpm for 1 min and the columns placed in clean 2 mL collection tubes. Again without wetting the rim, 500 μL of buffer AW2 (Qiagen) was added to the columns and these were centrifuged at 13,000 rpm for 3 min and the columns were placed in clean tubes. These were spun for 1 min at 13000 rpm to remove excess buffers and placed in clean collection tubes. Lastly, 200 μL of buffer AE (Qiagen) was added to the QIAamp spin columns and incubated at room temperature for 5 min and then centrifuged at 8000 rpm for 1 min. The last step was repeated and the DNA thus extracted as second sample.
Amplification of DNA
A 1500 bp fragment spanning COI to COII was amplified in a single reaction using the general insect primers ‘Jerry’ and ‘Pat’ (Simon et al., 1994) and ‘Muscid’ and ‘S2792’ (41) (Table 2). PCR amplification was carried out in a Perkin-Elmer 9700 Automated Thermocycler.
Sequencing
For sequencing, PCR products were purified using the Genclean for PCR Turbo Kit (Bio101). Each sample was sequenced in both forward and reverse directions using the ABI prism ® BigDye Terminator Cycle Sequencing Ready Reaction Kit (Perkin Elmer Applied Biosystems). This kit was diluted to produce a master mix containing two parts sequencing kit: 1 part 5x ABI Perkin-Elmer buffer ®: 1 part sterile water (Chippindale et al., 1998). Two microlitres of DNA, 2 μL of the sequencing mix and 1 μL of either forward or reverse primers at 1.6 pmol μL-1 concentration were then used in the reaction. The sequencing PCR program was carried out as follows: 25 cycles of 96°C for 10 sec, 50°C for 5 sec and 60°C for 4 min. Each individual was sequenced using the ‘Jerry’, ‘Muscid’ and the internal ‘Pat’ primers.
Sequence Alignment and Phylogenetic Analysis
Sequences were edited and aligned using the program Sequencer v3.1.2 (Gene codes) and translated into amino acid codons using the Drosophila genetic code option in the program. The reading frame of COI and COII was obtained by aligning existing Chalcidoid wasp sequences from GENBANK with the test sequences. The species chosen were Ceratosolen capensis (Grandi) (Hymenoptera: Agaonidae) (AF200377) and C. bisulcatus (AF200375) (Weiblen, 2001) and were used as outgroups for the subsequent analysis. In order to correctly align the tRNALEU sequences, the secondary structure was determined (Fig. 1) and alignment gaps placed in some sequences to preserve base pairing and accommodate insertion/deletions which are common in RNA sequences (Kjer, 1995). The use of secondary structure models greatly improves the accuracy of alignment and allows a more precise determination of homologous characters for phylogenetic analysis (Medina and Walsh, 2000). An existing arthropod tRNALEU structure was used as a template for alignment (Nardi et al., 2001). Stems were required to contain a minimum of two paired bases.
In order to correctly align the different genes between species and genera, alignment gaps were inserted between the 3’ end of COI and the 5’ end of the tRNA to accommodate a variable length non-transcribed region occurring between these two genes in some species. This phenomenon has also been described in other arthropod species (Stauffer, 1997; Weiblen, 2001). This non-transcribed region accounted for 91 bp and was excluded from the subsequent analyses.
 | |
| Fig. 1: | Alignment of tRNALEU to show secondary structure. Shaded area indicate areas of complimentary base pairing in strems |
| Table 3: | Percentage base composition between British Chalcid wasp genera |
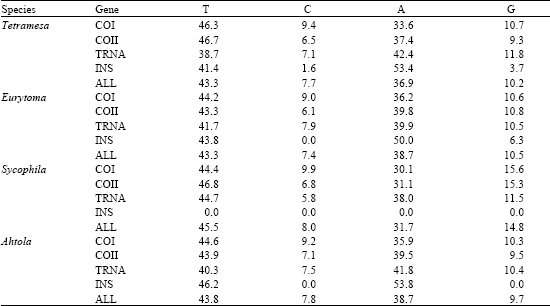 | |
Base composition (Table 3) and transition: transversion ratios (Table 4) were calculated in MEGAv2 (Kumar et al., 2001).
Phylogenetic analysis was conducted using PAUP 4.0b10 (Swofford, 2002). Gaps were treated as a 5th character state in all analyses. Neighbour-joining (NJ) and Maximum Likelihood (ML) methods were used for phylogenetic reconstruction.
| Table 4: | Transition:transversion ratios (R) and the number of transitions (s) and transversions (v) in pairwise comparisons of British Chalcid wasps |
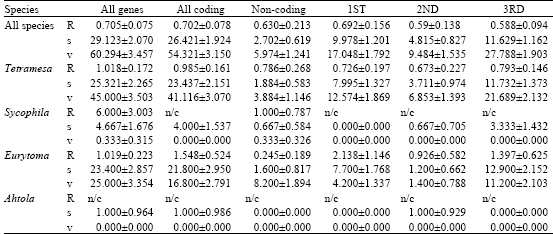 | |
Distance trees were derived using uncorrected p-values with the heuristic search and 1000 bootstrap replications. As an extreme A-T bias and saturated transitions have been reported in several species of fig wasp (family Agaonidae and Toryminae) mtDNA (Machado, 1998; Weiblen, 2001), neighbour-joining trees were also constructed using all substitutions and with transversions only, to determine whether saturation had an effect on tree topology.
The evolutionary model of ‘best fit’ for ML analysis was ascertained by running likelihood ratio test scores through Model test v3.06 (Posada and Crandall, 1998) with and without the non-transcribed sequence between COI and tRNALEU. In both cases, the best evolutionary model chosen was the ‘General Time Reversible’ (GTR)+G model. The shape of the gamma parameter (0.29) and the rate matrix were determined using PAUP and were employed using this model in subsequent heuristic searches to obtain the best ML trees for the data set.
RESULTS
Sequence Data
Complete DNA sequences for the region used in the analysis (551 bp) were obtained for 39 individuals from the four genera under study (Table 1). Base composition is shown in Table 3. Of the 546 bp sequence examined, 366 sites were potentially parsimony informative, although when the non-transcribed region was removed, this figure was reduced to 158 of 455 sites. Overall, the transition: transversion ratios were low (<1.0); this significant transversion bias possibly indicates saturation of multiple substitutions at the same site (Table 4).
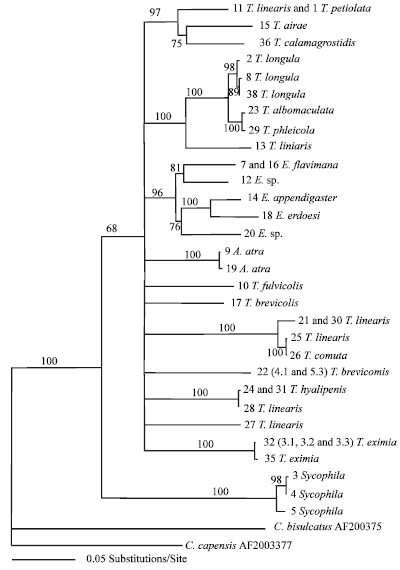
Distance based trees using all substitutions (Fig. 2) and those considering transversions only (Fig. 3), exhibited similar topologies. Using only transversions resulted in a tree which had better supported basal nodes than when all substitutions were considered, but support for internal branches was strong using both methods. The groupings of Ahtola, Sycophila and Eurytoma were strongly supported and distinct from each other and from Tetramesa. The types of substitution used for generating the tree did not affect this separation.
Using the GTR+G model, the -log likelihood score was 4325.2. Bootstrap analysis of the ML tree indicated good support for the Sycophila (100%), Ahtola (86%), but was less strong for Eurytoma (87%). Tetramesa clades were also well supported, although the relative position in the tree of three of the clades was not resolved (Fig. 4).
 | |
| Fig. 2: | NJ tree using all substitutions with 1000 bootstrap replications |
DISCUSSION
Molecular Evolution
By reporting the presence of a variable length non-transcribed region between the 3’ end of COI and the 5’ end of the tRNALEU in the four genera analysed here, this study confirms previous work in chalcid wasps (i.e., Agaonid fig wasps; Weiblen, 2001). To extend COI-COII analysis to other Chalcidoidea families is important in order to determine if this characteristic is common to the entire superfamily. A similar trait was described in a scolytid beetle (Stauffer, 1997), but the fact that such a feature is absent in the majority of the insect orders screened to date for this sequence fragment argues for a scenario of independent and incidental acquisition. A low transition bias or a tranversion bias in among-species comparisons suggests either that species are ancient or that rapid molecular evolution is taking place.
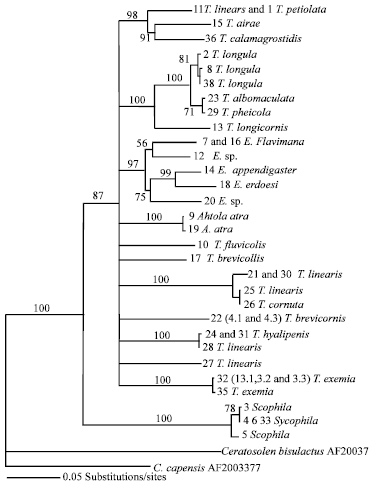 | |
| Fig. 3: | NJ tree using only transversions with 1000 bootstrap replications |
Since a transversion bias is typical of parasitic hymenoptera (Dawton and Austin, 1997) and not for parasitic dipterans (Castero et al., 2002), this feature does not seem to be associated with lifestyle nor probably with evolutionary age, but rather, with an overall accelerated evolutionary rate in Hymenoptera.
Phylogenetics and Taxonomy
The number of unambiguous or good quality sequences retrieved was some what limited compared with the total number of samples amplified for the target DNA fragment. The reason for this low success rate may be the existence of copies of COI-COII inserted in the nuclear genome (Numts) (Bensasson et al., 2001) that prevent straightforward sequencing of the true mitochondrial version of the gene. It was not possible to solve this problem by extensive cloning in the time available. However, the problem of Numts leading to spurious PCR amplifications will be one of the priorities of any extension of the research initiated here.
Although less of a problem for Tetramesa sp., from which sequences were obtained were obtained from fourteen different species, even so the sequence data was hindered by the possible presence of Numts in all the genera studied. This was particularly true for Sycophila to such an extent in fact that sequencing problems prevented the acquisition of significant data at a population level for the S. mellea complex.
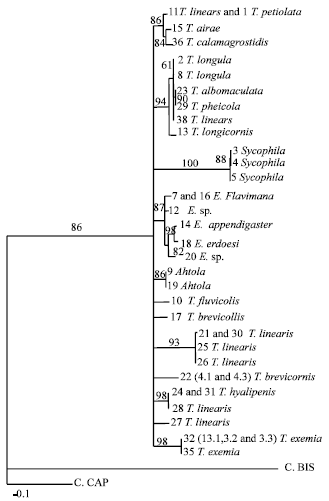 | |
| Fig. 4: | Maximum likehood tree using GTR+G following 100 bootstrap replications |
Even with the recognition that the data should be enlarged, both in terms of the total number of specimens screened and of Eurytoma and Sycophila species/populations studied, the monophyly of three of the four eurytomid genera is well supported by the different methods of phylogenetic reconstruction here applied to the sequence data. Consequently, the validity of Ahtola as a separate taxa is also shown from this analysis. Only Tetramesa, for which we have many more sequences than for other species, appears to be paraphyletic. In the case of the ML tree reconstructions, Eurytoma, Sycophila and Ahtola are monophyletic within the paraphyletic structure of Tetramesa, whereas in the NJ trees Sycophila is basal to the three other genera.
Several taxonomists have studied Tetramesa species (Al-Barrak et al., 2004; Claridge and Dawah, 1994; Dawah et al., 1995; Dawah, 1987). Despite the efforts of such researches, many Tetramesa species remain difficult to identify because of their uniform morphology. Molecular techniques could be useful tools to clear up their taxonomic status. For example, Al-Barrak et al. (2004) could successfully discriminated closely-related species of Tetramesa by using RAPD-PCR.
In terms of the within-genus taxonomy of Tetramesa, it is possible to draw some conclusions from the available results. It is interesting that the clades produced from the molecular phylogenies show some parallels with the trophic clusters of Tetramesa species associated with host grasses in the eurytomid food web (Dawah et al., 1995). A cluster is present with T. petiolata and T. airae (both herbivores of D. cespitosa) and two clusters occur containing all species that use E. repens. Furthermore, with the exception of T. linearis, all Tetramesa species included in the study seem to be paraphyletic and are therefore likely to be valid species, a result that extends previous findings using protein electrophoresis (Dawah, 1987). Al-Barrak et al., (2004) could discrimination between three closely-related species of T. calamagrostidis, T. longicornis and T. petiolata using RAPD-PCR. They reported that two forms of T. hyalipennis (ex: E. repens and E. farctus) were the most closely-related of any of the species investigated and probably diverged the most recently. They believed that some degree of sympatric evolution has occurred, most obviously in the case of the host adapted forms of T. hyalipennis. Dawah (1986) used electrophoresis for phylogenetic analysis of certain Tetramesa spp. And reported that T. hyalipennis reared from E. repens and E. farctus (Viv) represented a single species. He also stated that T. exemia reared from Calamagrostidis epigejos and Ammophila arenaria represented a single species which showed that there was a restriction of gene flow between these particular populations. Henneicke et al. (1992) used a comparative morphological study on final-instar larval stage of 33 species of grass-inhabiting wasps belonging to four genera: Eurytoma. Tetramesa, Sycophila and Ahtola. They found that Ahtola was intermediate between Eurytoma and Tetramesa and S. mellea was confirmed as a distinct genus.
DNA sequencing has been shown in this study to be a powerful tool which has allowed successful discrimination between the four eurtytomid genera, Tetramesa, Eurytoma, Ahtola and Sycophila. With the sequence data, the monophyletic status of three of the four genera is well supported by different methods of phylogenetic reconstruction. Only Tetramesa, which is by far the most numerous taxon here studied, seems paraphyletic but even in this case, there are certain phylogenetic relationships within the group that are interesting from both a morphological and host preference point of view. Thus for example, the T. petiolata and T. airae group together, which are both herbivores of D. cespitosa and two clusters occurred containing all species that use E. repens, i.e., T. linearis, T. hyalipennis and T. cornuta. Another interesting result of this analysis is the support for the validity of Ahtola as a separate taxon. Thus, these results clearly support the taxonomic separation of these four genera of eurytomids.
ACKNOWLEDGEMENT
I would like to thank to Tehran University and the Iranian Ministry of higher education for the award of a Ph.D Scholarship.
REFERENCES
- Avise, J.C., J. Arnold, R.M. Ball, E. Bermingham and T. Lamb et al., 1987. Intraspecific phylogeography: The mitochondrial DNA bridge between population genetics and systematics. Annu. Rev. Ecol. Syst., 18: 489-522.
CrossRefDirect Link - Dawah, H.A., B.A. Hawkins and M.F. Claridge, 1995. Structure of the parasitoid communities of grass-feeding chalcid wasps. J. Anim. Ecol., 64: 708-720.
Direct Link - Kumar, S., K. Tamura, I.B. Jakobsen and M. Nei, 2001. MEGA2: Molecular evolutionary genetics analysis software. Bioinformatics, 17: 1244-1245.
CrossRefPubMedDirect Link - Moritz, C., 1994. Applications of mitochondrial DNA analysis in conservation: A critical review. Mol. Ecol., 3: 401-411.
CrossRef - Moritz, C., T.E. Dowling and W.M. Brown, 1987. Evolution of animal mitochondrial DNA: Relevance for population biology and systematics. Annu. Rev. Ecol. Syst., 18: 269-292.
CrossRefDirect Link - Simon, C., F. Frati, A. Beckenbach, B. Crespi, H. Liu and P. Flook, 1994. Evolution, weighting and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am., 87: 651-701.
Direct Link - Stauffer, C., 1997. A Molecular Method for Differentiating Sibling Species Within the Genus Ips. In: Proceedings: Integrated Cultural Tactics into the Management of Bark Beetle and Reforestation Pests. Gregoire, J.C., A.M. Liebhold, F.M. Stephen, K.R. Day and S.M. Salom (Eds.) USDA Forest Service General Technical Report NE -236, pp: 87- 91.
- Villablanca, F.X., 1994. Spatial and Temporal Aspects of Populations Revealed by Mitochondrial DNA. In: Ancient DNA: Recovery and Analysis of Genetic Material from Palaeontological, Archaeological, Museum, Medical and Forensic Specimens, Herrmann, B. and S. Hummel (Eds.). Springer-Verlag, New York, pp: 31-58.