
Mohsen Ranjbar
Department of Medicinal Chemistry, Faculty of Pharmacy, Shiraz University of Medical Sciences, Shiraz, Iran
LiveDNA: 98.30889
Hamidreza Ghafouri
Department of Medicinal Chemistry, Faculty of Pharmacy, Shiraz University of Medical Sciences, Shiraz, Iran
Farnaz Salehi
Department of Medicinal Chemistry, Faculty of Pharmacy, Shiraz University of Medical Sciences, Shiraz, Iran
Leila Emami
Department of Medicinal Chemistry, Faculty of Pharmacy, Shiraz University of Medical Sciences, Shiraz, Iran
Neda Khonya
Department of Medicinal Chemistry, Faculty of Pharmacy, Shiraz University of Medical Sciences, Shiraz, Iran
Zahra Rezaei
Department of Medicinal Chemistry, Faculty of Pharmacy, Shiraz University of Medical Sciences, Shiraz, Iran
Amirhossein Sakhteman
Department of Medicine, Faculty of Health Sciences, University of Eastern Finland, Kuopio, Finland
LiveDNA: 98.30944
Journal of Biological Sciences
Year: 2020 | Volume: 20 | Issue: 1 | Page No.: 22-31







ABSTRACT
Background and Objective: Human histamine-2 (H2) receptor is a G-protein coupled type receptor which is one of the main targets of several therapeutics used in acid peptic disorders of the gastrointestinal tract. To perform more computational drug design studies on this receptor, it is incumbent to obtain more structural information about this protein. A model obtained based on the computational studies would provide a valuable tool for further structure-based drug design projects on H2 receptor which are inexpensive but profitable works. Materials and Methods: In this study, homology modeling studies and molecular dynamics simulation were done for the H2 receptor by using a DPPC lipid bilayer for 50 ns. Several frames of the simulated receptor were elicited based on simulation orientations exhibiting the receptor at different states. Cross-docking simulations of some inhibitors with known experimental values (Ki) have done to find an acceptable model of the protein at the antagonist state. Results: Frame 126 revealed a rational correlation between docking gained energy scores and experimental activity values (R = 0.9). It was the most reliable gained model of the protein. Conclusion: The obtained template of H2 receptor is practical enough to be entered into further computational studies. Reliability of the model has been approved through our docking studies and molecular dynamics simulation.
PDF Abstract XML References Citation
Copyright: © 2020. This is an open access article distributed under the terms of the creative commons attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
How to cite this article
Mohsen Ranjbar, Hamidreza Ghafouri, Farnaz Salehi, Leila Emami, Neda Khonya, Zahra Rezaei and Amirhossein Sakhteman, 2020. Homology Modeling, Molecular Dynamics Simulation and Cross-Docking Studies of Human Histamine-2 (H2) Receptor to Obtain a 3D Structure for Further SBDD Studies. Journal of Biological Sciences, 20: 22-31.
DOI: 10.3923/jbs.2020.22.31
URL: https://scialert.net/abstract/?doi=jbs.2020.22.31
DOI: 10.3923/jbs.2020.22.31
URL: https://scialert.net/abstract/?doi=jbs.2020.22.31
INTRODUCTION
G-protein coupled receptors (GPCRs) are a group of membrane receptors that are among the most diverse and biggest protein families inside the human body1. Approximately 2% of the human genome is related to these proteins2. Today, about 35% of the current drugs target is GPCRs. However, a little number of GPCRs are aimed as therapeutic goals at the present time3.
Histamine receptors are among the most noticeable types of GPCRs. The receptors of this family are widely spread through the organs. Four types of these receptors are recognized: H1-H4. The concentration of the current study is on the H2 receptors. H2 receptors are known as noticeable modulators of the gastric system. These receptors have an important role in gastric acid secretion. The H2 receptor antagonists are among the most applied drugs in gastric disorders. In addition, the activation of the H2 receptor results in sinus rhythm increase and smooth muscle relaxation. H2 receptor is a kind of GPCR which its activation increases the cAMP level drastically4.
Currently, there is no proper 3D structure for the H2 receptor (based on the Protein Data Bank, www.rcsb.org). This limitation hampers the computational studies on this receptor. In this study, the main goal is to obtain an appropriate model for the H2 receptor, based on homology modeling. Molecular dynamics simulations and ligand-protein interaction studies are useful techniques to evaluate the validity of the modeling5,6.
A significant impediment for computational studies on the H2 receptor is the absence of an appropriate 3D structure for this receptor. A 3D model of the human histamine H1 receptor has been industrialized by homology. Usage of docking calculations to find the role of amino acids affecting agonist or antagonist binding was performed based on the genetic algorithm7. In another study on H2 receptor, cimetidine, ranitidine and nizatidine as antagonists were used to defining binding sites with a ligand by molecular docking, molecular dynamics simulations. One aspartic acid, Asp98 in transmembrane domain 7 (TM3), has been identified as main suppliers to ligand binding with H-bond interactions. Besides, Asn159 in TM4 and Asp186 in TM5 have important roles in the stabilizing complex of H2-antagonist8. A review article shows the importance of homology modeling in defined recent improvements and predicting protein structure with successful applications at the levels of the drug design and discovery9.
Structure-based drug design, which is a more-prominent technique than the ligand-based method, needs 3D conformations of the proteins. Agonist and antagonist states of a protein are 2 considerable applications for structure-based drug design studies10,11. The goal of this study is to extract a model of the receptor using current available 3D templates. For this purpose, homology modeling, which has been described as a robust computational technique to predict the structure of the transmembrane proteins such as GPCRs, has been utilized in this research5,12,13. In the following, to achieve the orientations of the receptor, molecular dynamics (MD) simulation studies have been performed. As a normal work in GPCRs studies, the structure of the protein has been assigned into a lipid bilayer to imitate physiological conditions. Finally, the extracted frames of the receptor and current known structures with available experimental activity (Ki) have gone through cross-docking studies. To achieve the most applicable frame for structure-based drug design studies (SBDD), one with the most correlation between experimental and gathered computational data has been selected.
MATERIALS AND METHODS
Study area: This study was conducted at the Pharmacy School, Shiraz University of Medical Sciences, Iran. This project has been started in January, 2019 and ended in May, 2019.
Research procedure: A core i7 laptop on windows 8.1 was utilized for the preparation chemical structure of the compounds. Simulation processes were run by a 24 core computational server on Linux Ubuntu12. First, for the purpose of the homology modeling process, the UniProt database (www.uniprot.org) was used for drawing out the sequence of H2 receptor (PDB code:P25021) as a FASTA template14. I-TASSER search engine (Iterative Threading ASSEmbly Refinement http://zhanglab.ccmb. med.umich.edu/ I-TASSER) determined the acquired sequence to recognize the templates from the Protein Data Bank.
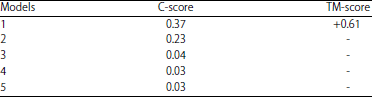
The highest C-score obtained from the I-TASSER server was chosen as the best PDB structure to run the MD simulation process15.
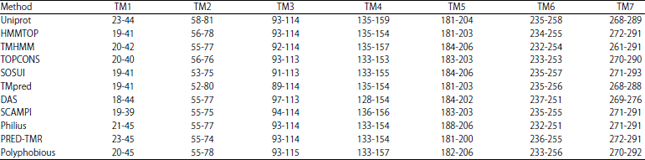
In this study, 11 servers that mentioned below were utilized to predict transmembrane helices or alignment the model that possesses an accurate topology inside lipid bilayer:
| • | TOPCONS (http://topcons.cbr.su.se)16 |
| • | HMMTOP (http://www.enzim.hu/hmmtop)17 |
| • | DAS (http://www.enzim.hu/DAS/DAS.html)18 |
| • | SOSUI (http://harrier.nagahama-i-bio.ac.jp/sosui)19 |
| • | TMHMM (http://www.cbs.dtu.dk/services/TMHMM/) |
| • | TMpred (http://www.ch.embnet.org/ software/TMPRED _form.html)20 |
| • | PolyPhobius (http://phobius.sbc.su.se/poly.html) |
| • | SCAMPI (https://omictools.com/scampi-tool) |
| • | PREDTMR(http://athina.biol.uoa.gr/PREDTMR/) |
| • | Philius(https://omictools.com/philius-tool) |
| • | UniProt (https://www.uniprot.org/help/transmem) |
In the next step, to get started molecular dynamics simulation, GROMOS96 53A6 force field was applied as performed in Gromacs 4.5.5 that describes the lipids derived by Berger lipid parameters21 and situates the receptor inside a lipid bilayer. A 128 DPPC (dipalmitoyl-phosphatidylcholine) and some commands are needed for the simulation. Also, VMD software was employed to align the intended procedure22.
A method called the Inflate GRO develops the system totally for the elimination of extra lipid residues and incorporation of the protein23. To set the protein in the bilayer membrane, the mentioned method was employed in the role of an algorithm. In the next step, the most appropriate area per lipid for DPPC systems was prepared by repetitive runs of shrinking and minimization24.
Area per lipid density during all steps of shrinking and minimization was calculated by employing GridMAT-MD_v2.0 Perl script25. Following this, water and ions were added to the simulating system in order to deter permeation of water molecules inside the hydrophobic parts of the lipid membrane. In this step, the van der Waals radius of carbon atom was set to 0.375Å. A concentration of 0.15M NaCl was added on the system to simulate the physiological environment. After the Energy minimization step, the protein backbone was restrained. The simulation followed by 2 equilibration experiments including NVT and NPT ensemble. In these steps the system was subjected to a minimization step. While the Nose-Hover algorithm was used as an accurate thermostat in the NPT ensemble, a modified Berendsen was used in the NVT v-scale. Periodic Boundary Condition (PBC) was prepared during the simulation by using Particle Mesh Ewald (PME)long-range electrostatics. A 50 ns Molecular Dynamics Simulation was executed on the system to give enough time for conformational changes inside the receptor. In order to begin the docking process, the receptor should be sampled. For a sampling of several receptor conformations, TCL scripting was employed. Therefore, 200 PDB structures were adapted from the output file of MD trajectories covering all simulation time. Via assigning Gasteiger partial charges using MGLTOOLs 1.5.6 the obtained PDB files were converted to pdbqt files26. In the next step, to prepare ligand, a collection of 28 molecule ligands were resumed from the CHEMBL database27. In the next step, by using Open babel 2.3.2 the structures were converted to mol2 format. By the way, the Ki values were retrieved and saved into PKi (-logki). MGLTOOLS 1.5.6, for production 28 pdbqt files, was used to add the Gasteiger partial charges and merge non-polar hydrogen atoms.
Afterward, for the determination of the binding site, the sequence of H2 receptor was subjected to RaptorX(http:// raptorx.uchicago.edu)28.
Finally, in order to carry out the Molecular Docking process coordinate and size of the grid box must have been determined in advance. Based on the former literature, Coordinates of Cα for LYS121 were assumed as the grid box center. The size of the grid box computed according to the equation below:
Size x; y; z = 2×LDA = 30Å
LAD marks the spot that atomic distance is the largest in all data collections. Afterward, the AutoDock Vina 1.1.2 software25 was utilized for 200*33 = 6600 cross-docking simulations29. Exhaustiveness value was set to 100 to execute impressive docking simulations in AutoDock Vina for all outputs.
Statistical analysis: The Pearson correlation coefficient (R) as the statistical tool for analyzing the correlation of docking results, was computed between every single obtained docking energy value and the pKi values for each frame. Receptor-ligand interactions were shown based on some pattern gotten by protein-ligand interaction profiler (PLIP) (https://projects. biotec.tu-dresden.de/plip-web/plip/)30.
RESULTS
Five templates were used during the modeling: 3MY9_A, 4U16_B, 32PQ_A, 2Y01_A and 2Y00_B.
The top 5 I-TASSER models based on their C-score is represented in Table 1. The model with the highest C-score was considered as the best one.
| Table 1: | Top 5 I-TASSER models based on their C-score |
 | |
| Table 2: | Eleven methods were applied in order to predict TM regions of H2-receptor |
 | |
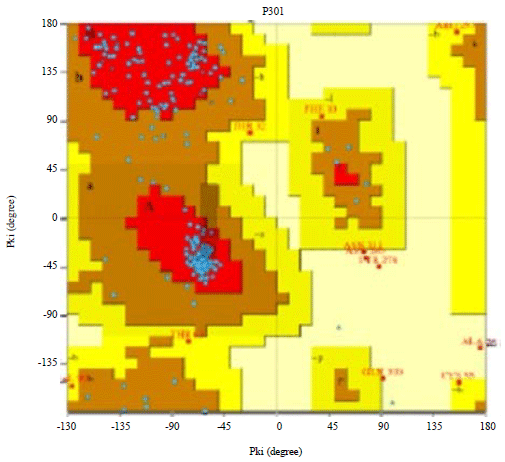 | |
| Fig. 1: | Ramachandran plot for the protein was obtained from PROCHECK |
The TM-score for the bestmodel is higher than 0.5 which means it has an acceptable topology for further modeling studies.
Ramachandran plot for the protein was obtained from PROCHECK Server (Fig. 1)31. The function of this plot is to verify the obtained model. More than 98% of the residues are placed in the favorable part of the plot, which means that conformational features of the modeled protein are tantamount to the native proteins.
11 methods were applied in order to predict TM regions of H2-receptor. As presented in Table 2, the topology of membrane proteins has been predicted precisely and efficiently.
After obtaining an acceptable model for the 3D structure of the protein, the next step was to perform molecular dynamics (MD) simulations. The purpose of these simulations was to recognize the various conformations of the protein in the physiological conditions. A 50 ns MD simulation was conducted on the whole system to find out an equilibrate state for the receptor based on the RMSD variation of Cα and energy plot. The result of this step is depicted in Fig. 2a-b. The steady-state reaching time was 44 ns after the beginning of the process. Additionally, the energy diagram shows an equilibration of energy during the simulation.
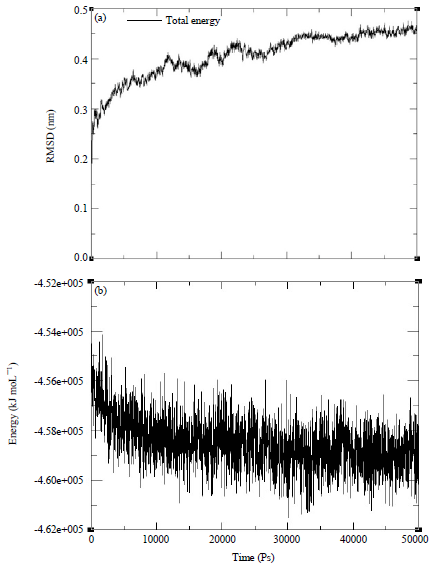 | |
| Fig. 2(a-b): | A 50 ns MD simulation was conducted on the whole system to find out an equilibrate state for the receptor based on (a) RMSD variation of Cα and (b) Energy plot |
To find out the most fluctuating parts of the receptor, a heat-map plot was delineated using the VMD software. This plot is shown in Fig. 3. Based on the heat-map, two extracellular domains of the protein showed a high level of fluctuation. These two domains are the residues 1-10 and 161-180. Additionally, a cytoplasmic domain revealed a high degree of fluctuation. This was the domain containing the residues 290-311.
For the docking simulations, it was necessary to find out the central residue of the grid box. Based on the RaptorX method and using of MOE, LYS121 was selected as the center. In order to opt the best frame for the docking simulation, the Pearson correlation of all docking frames with PKi values was attained. The most correlation was calculated for the frame 126 (|r| = 0.9) (Fig. 4).
To provide an outline for the binding mode of the ligands in the active site of H2-receptor, the interaction maps of the receptor for all the compounds were obtained. The results of all ligand-receptor interactions have been represented in Table 3.
CHEMBL474991 has the best docking score (-10.1 kcal moL–1). Figure 5 depicts the interactions between CHEMBL474991 and the active site of the target.
Mobility of binding site residues was also calculated by mapping the RMSD values of the relevant residues during the simulation. As illustrated in Fig. 6 variations of the binding site was fixed at the last steps of simulation with an RMSD average of 2Å.
DISCUSSION
Obtained results of this study shows a reliable template for human histamine-2 receptor. This 3D template has been verified by molecular dynamics simulation and cross-docking evaluation.
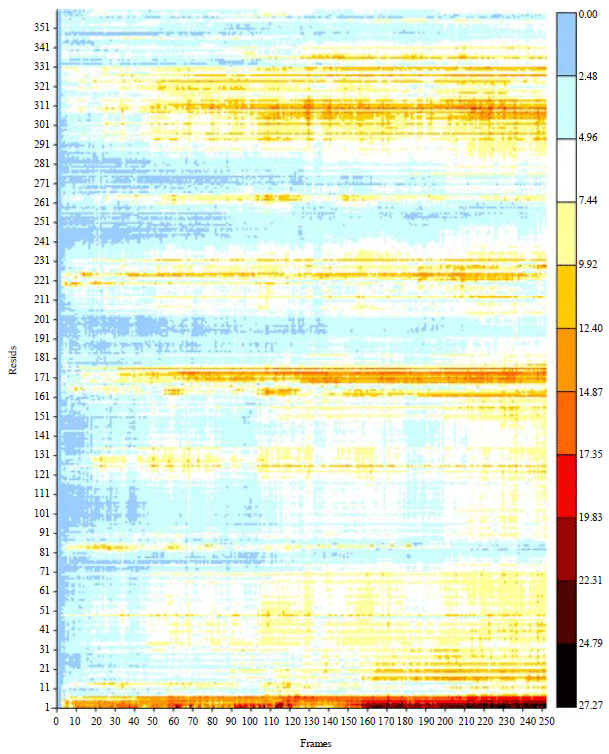 | |
| Fig. 3: | To find out the most fluctuating parts of the receptor, a heat-map plot was calculated using the VMD software |
The original technique used in this study is homology modeling. Nowadays, homology modeling plays a major role in all levels of computational biology: from genes to macromolecules32. It can be called the most precise method for structural prediction6. It is also a beneficial tools for more comprehensive studies such as pathway detections33. Homology modeling for proteins is not confined to finding the structure of receptors. it make us able to design more confident peptide drugs such as antibodies34. Structure-based drug design requisite is a comprehensive library of receptors in human body. However, our current treasure of proteins is not as exhaustive as needed. Homology modeling can play a benevolent role in empowering this collection. As some previous studies have proven, homology modeling alongside molecular dynamics and docking simulations can be an inexpensive way to shed the light on the unknown structures5,35. The output of this study can be seemed as an inchoate achievement.
| Table 3: | All ligand-receptor interaction |
 | |
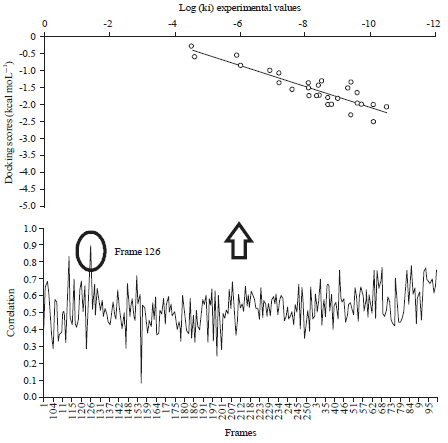 | |
| Fig. 4: | In order to opt the best frame for the docking simulation, Pearson correlation of all docking frames with Pki values were attained |
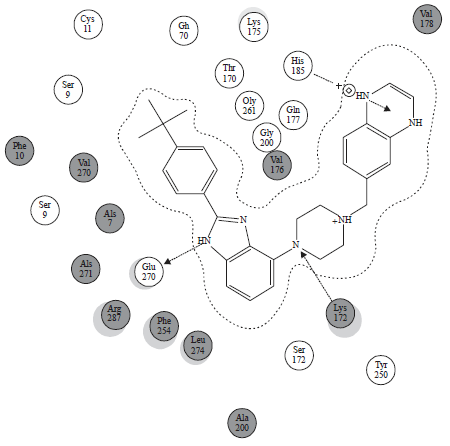 | |
| Fig. 5: | Interactions between CHEMBL474991 and the active site of the target |
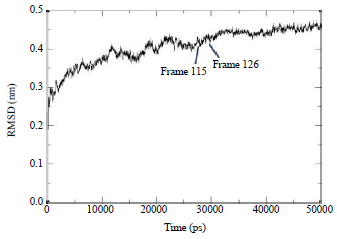 | |
| Fig. 6: | Mobility of binding site residues was also calculated by mapping the RMSD values of the relevant residues during simulation |
This template can be recruited by further computational studies on H2 receptor to become a definitely approved model.
CONCLUSION
The validity of obtained template for H2 receptor was testified by means of several in silico techniques. A high level of similarity between the drawn model and the predicted structure was established by a Ramachandran plot. Cross-docking studies for recognized H2 receptor antagonists were our second mechanism to justify the obtained model. These studies were performed on the frame 126 which had extracted by molecular dynamics simulations. According to the Pearson correlation test, one frame was accepted. The coefficient for this frame was 0.9 that represents a high degree of reliability for the results. Furthermore, our computational method could perfectly mimic the human H2 receptor at the antagonist state.
SIGNIFICANCE STATEMENT
This research plays a crucial role in the prospective drug design studies in order to recognize new ligands. Due to the unavailability of the protein structure of H2 receptor, the findings of this study pave the way for prospective computational studies on this receptor. The achieved structure based on this study enlarges the current library of human proteins, which is a strategic database for biological researches. Additionally, this study affirms the viability of homology modeling as an accessible, affordable and valuable technique in biological studies. Further unavailable protein structure can be designed by means of the methods utilized in the current study.
ACKNOWLEDGMENT
This research project was conducted from January-May, 2019. This Research Project was fully sponsored by Shiraz University of Medical Sciences with grant number 8915574058.
REFERENCES
- Venter, I.C., M.D. Adams, E.W. Myers, P.W. Li and R.J. Mural et al., 2001. The sequence of the human genome. Science, 291: 1304-1351.
CrossRefDirect Link - Fredriksson, R., M.C. Lagerstrom, L.G. Lundin and H.B. Schioth, 2003. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups and fingerprints. Mol. Pharmacol., 63: 1256-1272.
CrossRefDirect Link - Sriram, K. and P.A. Insel, 2018. G protein-coupled receptors as targets for approved drugs: How many targets and how many drugs? Mol. Pharmacol., 93: 251-258.
CrossRefDirect Link - Sadek, B. and H. Stark, 2016. Cherry-picked ligands at histamine receptor subtypes. Neuropharmacology, 106: 56-73.
CrossRefDirect Link - Khoddami, M., H. Nadri, A. Moradi and A. Sakhteman, 2015. Homology modeling, molecular dynamic simulation and docking based binding site analysis of human dopamine (D4) receptor. J. Mol. Model., Vol. 21, No. 2.
CrossRefDirect Link - Muhammed, M.T. and E. Aki-Yalcin, 2019. Homology modeling in drug discovery: Overview, current applications and future perspectives. Chem. Biol. Drug Design, 93: 12-20.
CrossRefDirect Link - Kiss, R., Z. Kovári and G.M. Keseru, 2004. Homology modelling and binding site mapping of the human histamine H1 receptor. Eur. J. Med. Chem., 39: 959-967.
CrossRefDirect Link - Zhang, J., T. Qi and J. Wei, 2012. Homology modeling and antagonist binding site study of the human histamine H2 receptor. Med. Chem., 8: 1084-1092.
CrossRefDirect Link - Vyas, V.K., R.D. Ukawala, M. Ghate and C. Chintha, 2012. Homology modeling a fast tool for drug discovery: Current perspectives. Indian J. Pharm. Sci., 74: 1-17.
CrossRefPubMedDirect Link - Cavasotto, C.N. and S.S. Phatak, 2009. Homology modeling in drug discovery: Current trends and applications. Drug Discov. Today, 14: 676-683.
CrossRefDirect Link - Schmidt, T., A. Bergner and T. Schwede, 2014. Modelling three-dimensional protein structures for applications in drug design. Drug Discov. Today, 19: 890-897.
CrossRefDirect Link - Katritch, V., M. Rueda, P.C.H. Lam, M. Yeager and R. Abagyan, 2010. GPCR 3D homology models for ligand screening: Lessons learned from blind predictions of adenosine A2a receptor complex. Proteins, 78: 197-211.
CrossRefDirect Link - Franca, T.C.C., 2015. Homology modeling: An important tool for the drug discovery. J. Biomol. Struct. Dyn., 33: 1780-1793.
CrossRefDirect Link - Gasteiger, E., A. Gattiker, C. Hoogland, I. Ivanyi, R.D. Appel and A. Bairoch, 2003. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res., 31: 3784-3788.
CrossRefPubMedDirect Link - Zhang, Y., 2008. I-TASSER server for protein 3D structure prediction. BMC Bioinformat., Vol. 9.
CrossRef - Bernsel, A., H. Viklund, A. Hennerdal and A. Elofsson, 2009. TOPCONS: Consensus prediction of membrane protein topology. Nucleic Acids Res., 37: W465-W468.
CrossRefDirect Link - Tusnady, G.E. and I. Simon, 1998. Principles governing amino acid composition of integral membrane proteins: Application to topology prediction. J. Mol. Biol., 283: 489-506.
CrossRefDirect Link - Cserzo, M., E. Wallin, I. Simon, G. von Heijne and A. Elofsson, 1997. Prediction of transmembrane alpha-helices in prokaryotic membrane proteins: The dense alignment surface method. Protein Eng., 10: 673-676.
CrossRefPubMedDirect Link - Hirokawa, T., S. Boon-Chieng and S. Mitaku, 1998. SOSUI: Classification and secondary structure prediction system for membrane proteins. Bioinformatics, 14: 378-379.
CrossRefDirect Link - Hofmann, K. and W. Stoffel, 1993. TMbase-A database of membrane spanning protein segments. Biol. Chem. Hoppe-Seyler, 374: 166-166.
Direct Link - Pronk, S., S. Pall, R. Schulz, P. Larsson and P. Bjelkmar et al., 2013. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics, 29: 845-854.
CrossRefDirect Link - Humphrey, W., A. Dalke and K. Schulten, 1996. VMD: Visual molecular dynamics. J. Mol. Graph., 14: 33-38.
CrossRefDirect Link - Schmidt, T.H. and C. Kandt, 2012. LAMBADA and InflateGRO2: Efficient membrane alignment and insertion of membrane proteins for molecular dynamics simulations. J. Chem. Inform. Model., 52: 2657-2669.
CrossRefDirect Link - Kandt, C., W.L. Ash and D.P. Tieleman, 2007. Setting up and running molecular dynamics simulations of membrane proteins. Methods, 41: 475-488.
CrossRefDirect Link - Allen, W.J., J.A. Lemkul and D.R. Bevan, 2009. GridMAT-MD: A grid‐based membrane analysis tool for use with molecular dynamics. J. Comput. Chem., 30: 1952-1958.
CrossRefDirect Link - Morris, G.M., R. Huey and A.J. Olson, 2008. Using autodock for ligand-receptor docking. Curr. Protoc. Bioinf., 24: 8.14.1-8.14.40.
CrossRefDirect Link - Gaulton, A., L.J. Bellis, A.P. Bento, J. Chambers and M. Davies et al., 2012. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res., 40: D1100-D1107.
CrossRefDirect Link - Kallberg, M., H. Wang, S. Wang, J. Peng, Z. Wang, H. Lu and J. Xu, 2012. Template-based protein structure modeling using the RaptorX web server. Nat. Protocols, 7: 1511-1522.
CrossRefDirect Link - Trott, O. and A.J. Olson, 2010. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem., 31: 455-461.
CrossRefPubMedDirect Link - Salentin, S., S. Schreiber, V.J. Haupt, M.F. Adasme and M. Schroeder, 2015. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res., 43: W443-W447.
CrossRefDirect Link - Laskowski, R.A., M.W. MacArthur, D.S. Moss and J.M. Thornton, 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Applied Cryst., 26: 283-291.
CrossRefDirect Link - Watkins, A.M., R. Rangan and R. Das, 2019. Using Rosetta for RNA homology modeling. Methods Enzymol., 623: 177-207.
CrossRefDirect Link - Lekeux, G., J.M. Crowet, C. Nouet, M. Joris and A. Jadoul et al., 2018. Homology modeling and in vivo functional characterization of the zinc permeation pathway in a heavy metal P-type ATPase. J. Exp. Bot., 70: 329-341.
CrossRefDirect Link - Jetha, A., N. Thorsteinson, Y. Jmeian, A. Jeganathan, P. Giblin and J. Fransson, 2018. Homology modeling and structure-based design improve hydrophobic interaction chromatography behavior of integrin binding antibodies. MAbs, 10: 890-900.
CrossRefDirect Link - Sakhteman, A., M. Khoddami, M. Negahdaripour, A. Mehdizadeh, M. Tatar and Y. Ghasemi, 2016. Exploring 3D structure of human gonadotropin hormone receptor at antagonist state using homology modeling, molecular dynamic simulation and cross-docking studies. J. Mol. Model., Vol. 22, No. 9.
CrossRefDirect Link