
Socorrina Colaco
Department of Pharmacology, Srinivas Institute of Medical Sciences and Research Centre, Srinivas Nagar, Mukka, Surathkal, Mangalore, 575021, Karnataka, India
N. Ramesh
Department of Pharmaceutics, Srinivas College of Pharmacy, Farengipete Post, Mangalore, 574143, Karnataka, India
Sekar Rajan
Department of Pharmaceutical Analysis, J.S.S. College of Pharmacy, Rocklands, Ooty, 643001, Tamilnadu, India
Subramania Nainar Meyyanathan
Department of Pharmaceutical Analysis, J.S.S. College of Pharmacy, Rocklands, Ooty, 643001, Tamilnadu, India
Journal of Biological Sciences
Year: 2015 | Volume: 15 | Issue: 2 | Page No.: 102-105







ABSTRACT
Objective was to compare pharmacokinetic characteristics of Sustained-Release (SR) and Immediate-Release (IR) formulations of dextromethorphan hydrobromide following a single oral administration of test and reference formulations in fasting healthy male volunteers. A randomized, 3-way, crossover, bioequivalence study was conducted in 6 healthy male volunteers to compare dextromethorphan hydrobromide Sustained Release (SR) tablet as test and the Immediate Release (IR) as reference product. Blood samples were collected and plasma samples were determined by using validated HPLC method involving a solid phase extraction method. Pharmacokinetic parameters were calculated by non-compartmental analysis. Cmax of the sustained-release formulation was significantly lower than that of the marketed immediate-release. The Tmax of sustained-release formulation was significantly long-lasting than that of immediate release. These results are in line with the profile of a sustained-release drug. This was also evident by the lower elimination rate and higher t½ values. However, the bioavailability of sustained-release tablets remained the identical as that of immediate release tablets.
PDF Abstract XML References Citation
Received: June 09, 2015;
Accepted: August 29, 2015;
Published: September 19, 2015
How to cite this article
Socorrina Colaco, N. Ramesh, Sekar Rajan and Subramania Nainar Meyyanathan, 2015. A Single-Dose, Three-Period, Six-Sequence Crossover Study Comparing the Bioavailability Study of Dextromethorphan Hydrobromide Sustained Release (SR) Tablet and the Immediate Release (IR) Tablet in Healthy Volunteers Under Fasting Condition. Journal of Biological Sciences, 15: 102-105.
DOI: 10.3923/jbs.2015.102.105
URL: https://scialert.net/abstract/?doi=jbs.2015.102.105
DOI: 10.3923/jbs.2015.102.105
URL: https://scialert.net/abstract/?doi=jbs.2015.102.105
INTRODUCTION
Dextromethorphan hydrobromide is a synthetic antitussive compound which is frequently used with an antihistamine in treating unproductive cough. Dextromethorphan suppresses cough by central action on cough center in medulla. It has no analgesic or little sedative properties. Dextromethorphan is well absorbed from GI tract. It is metabolized in the liver and excreted in the urine as unchanged dextromethorphan and demethylated metabolites including dextrorphan, which has cough suppressant activity (Silvati et al., 1987). Numerous pharmacokinetic studies on dextromethorphan have been reported (Woodworth et al., 1987a, b; Demirbas et al., 1998; Ramachander et al., 1977; Silvasti et al., 1990; Yeh et al., 2003; Kukanich and Papich, 2004; Hu et al., 2011; Liu et al., 2004; Fossati et al., 1993; Kazis et al., 1996).
The objective of the present study was to conduct comparative bioavailability study of the dextromethorphan hydrobromide sustained release (slow and fast) and the marketed Immediate Release (IR) formulation in healthy male volunteers.
MATERIALS AND METHODS
Ethical conduct of study and informed consent: This study was conducted in compliance with the Harmonized Tripartite Guidelines for Good Clinical Practice (GCP) issued by the International Conference on Harmonization (ICH), the local laws and regulations (ICMR Guidelines on Biomedical Research, CDSCO) for the use of investigational therapeutic agents and the provisions of declaration of Helsinki. The Clinical Study Protocol was unconditionally approved by the ethics committee. A study-specific informed consent form was provided to each subject prior to drug administration, allowing sufficient time for review of the information provided and to ensure that subjects were aware of the implications of enrolling in the study prior to signing a study-specific form prior to period I. All subject identities were kept confidential.
Overall study design: This was an open-label, randomised, single-dose, three-way crossover, six-sequence, comparative bioavailability study performed on 6 healthy adult volunteers. A total of 6 subjects completed the clinical phase of the study. Prior to study commencement, subjects were randomly assigned to a treatment in accordance with the randomization scheme generated by Microsoft® Excel software. Subjects were confined to the CPU from at least 10 h prior to drug administration until after the 24.0 h post-dose blood draw, in each period. The treatment phases were separated by a washout period of 7 days.
Selection of study population: Subjects enrolled in this study were members of the community at large. Subject screening procedures included informed consent, inclusion/exclusion check, demography, medical history, medication history, physical examination, height, weight, body mass index, a concomitant medication check, vital signs measurements (blood pressure, pulse rate, respiratory rate and oral temperature), a 12-lead electrocardiogram (ECG), a urine drug screen, hematology, biochemistry, urinalysis, HIV and hepatitis testing. All participating subjects were judged eligible for the study when assessed against the inclusion and exclusion criteria.
Treatments administered: Subjects were administered a single oral dose of either the test dextromethorphan hydrobromide SR 60 mg tablets (slow, fast) or reference products (Romilar® tablets containing 15 mg of dextromethorphan). The treatments were administered with 240 mL of water.
Food and fluid intake: Subjects were served a controlled meal not less than 4 h post-dose and standard meals at appropriate times thereafter, in each period. Subjects were served identical post-dose meals in each period. With the exception of the volume administered at the time of dosing fluids were not permitted from 1 h before dosing to 1 h after dosing but water was permitted ad-libitum at all other times.
Restrictions: Subjects abstained from food or drink containing xanthine derivatives or xanthine-related compounds, alcohol-based products and energy drinks from 48 h prior to drug administration, until the end of sample collection in each period (24 h post-dose draw) and food or beverages containing grapefruit, natural food supplements (including St. John’s Wort or herbal products derived from St. John’s Wort-Hypericum Perforatum) and vitamins from 7 days prior to drug administration, until the end of sample collection in each period. Subjects did not smoke from at least 2 h prior to drug administration until 4 hours post-dose, in each period.
Physical activity: Subjects were required to remain seated in a semi-reclined position and avoid lying down or sleeping (unless medically necessary, procedurally required, or to go to the bathroom) for the first 2 h after dosing in each period. Vigorous physical activity was prohibited at all times during the confinements.
Pharmacokinetic sampling
Blood sample collection, processing and storage: A total of 26 (1×4 mL) samples of venous blood were obtained in each period. Samples were collected within one hour prior to drug administration (pre-dose) and at 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 4.0, 6.0, 8.0, 12.0, 18.0 and 24.0 h after drug administration in each period. The pre-dose and post-dose samples up to 12 h were collected by using disposable syringes via indwelling cannula placed in an ante-cubital vein or one of the forearm veins inserted prior to first sample collection in each period. The cannula left on the subject until the 12 h blood sample collection. Heparin lock technique was used to prevent the clotting of blood in the in-dwelling cannula. About 0.5 mL of the heparinized blood withdrawn and discarded prior to each scheduled blood draw. If there was more than 2 draws to be done after the cannula is blocked, a fresh cannula inserted, after removal of the blocked cannula. Actual time of collection of blood sample (to the nearest minute) noted. The collected blood transferred immediately after collection into pre-labelled vacutainer. Blood samples were cooled in an ice bath and were centrifuged at 3,500 rpm for at least 10 min at approximately 4°C. Plasma was dispensed into polypropylene tubes (as soon as possible). The aliquots were subsequently transferred to a -30°C freezer until pending analysis.
RESULTS
Calibration curve standards consisting of a set of eight non-zero concentrations ranging from 110.0, 140.0, 230.0, 590.0, 1000.0, 1400.0, 2100.0 and 3000.0 ng mL–1 for dextromethorphan were prepared. Quality control samples were prepared at 3 different concentrations of 110.0 ng mL–1 Quality Control-Lower Limit of Quantification (QC LLOQ), 590.0 ng mL–1 (QCL), 1400.0 ng mL–1 Quality Control- Medium (QCM) and 3000.0 ng mL–1 Quality Control-High (QCH). The lower limit of quantification was 110 ng mL–1. The method was validated over the range of 110.0-3000.0 ng mL–1. Correlation coefficient (r2) was 0.9984. Acceptable intra-day and inter-day precision (<15%) and accuracy (<10% diff.) were observed over the linear range of 110-3300 ng mL–1 as presented in Table 1: precision and accuracy for dextromethorphan. No significant interference was observed at the RT of dextromethorphan and internal standard in all the batches screened. The absence of any matrix effects was displayed. The percentage recovery of the analyte was 97.43%. The recovery of analyte was consistent at all levels.
The analyte was stable in human plasma for 3 days when stored below -50°C and for 5 h and 45 min when stored on bench top at room temperature. The analyte was stable till four freeze-thaw cycles. The analyte and internal standard in stock dilution and stock solution were stable. The validated technique found appropriate for sample analysis. The plasma samples were determined by a validated HPLC method. Sample preparation achieved by solid phase extraction method. Losartan potassium was used as Internal Standards (IS). Dextromethorphan and IS were extracted using Acetonitrile: 0.5% trifluoro acetic acid 40:60 and analyzed by using VYDAC Monomeric C18 (250×4.6 mm i.d., 5 μ).
The sustained release tablets were well absorbed and the extent of absorption was higher than that of the immediate release tablet.
| Table 1: | Precision and accuracy for dextromethorphan |
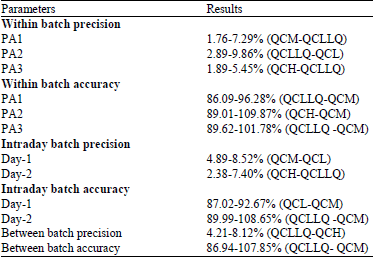 | |
The sustained and efficient drug delivery system developed in the contemporary study will maintain plasma levels better, which will overcome the drawbacks associated with the conservative therapy. After a single dose the Cmax of the sustained-release formulation was significantly lower than that of the marketed immediate-release. Tmax of sustained-release formulation was significantly long-lasting than that of immediate release as mentioned in Table 2. These results are in line with the profile of a sustained-release drug as in the Fig. 1. This was also evident by the lower elimination rate and higher t½ values. However, the bioavailability of sustained-release tablets remained the identical as that of immediate release tablets.
DISCUSSIONS
Several studies on dextromethorphan have been stated (Silvati et al., 1987, Silvasti et al., 1990; Woodworth et al., 1987a; Demirbas et al., 1998; Ramachander et al., 1977), all these studies have carried out in combination drug, none of these studies have carried out to demonstrate the once daily dosage of dextromethorphan in healthy volunteers.
Six healthy, adult, human male subjects planned and enrolled for the study as per the protocol. Six subjects completed both the periods. Pharmacokinetic analyses were performed on data obtained from six subjects who completed the study, as per the study protocol.
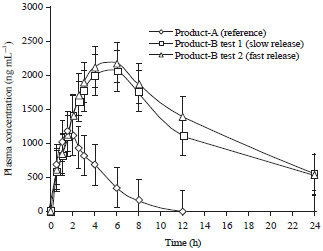 | |
| Fig. 1: | Plasma concentration-time profile of dextromethorphan hydrobromide from developed sustained release tablets (test) and marketed immediate release tablet |
| Table 2: | Mean pharmacokinetic parameters of the developed sustained release tablet and marketed immediate release tablets of dextromethorphan hydrobromide |
 | |
A validated HPLC method was used for the estimation of dextromethorphan in human plasma from the clinical samples of the current project. The analyte was extracted using a solid phase extraction procedure. The Lower Limit of Quantization (LLOQ) was 110.0 ng mL–1 and the Upper Limit of Quantization (ULOQ) was 3000.0 ng mL–1. This report provides the results of back calculated calibration curve standards data, quality control samples and study sample data.
The measured concentrations for each subject for all the time points were calculated against the calibration curve prepared with known standards. Concentrations below the BLQ were set to ‘zero’. The pharmacokinetic parameters (Primary parameters: Cmax, AUC last and AUCinf and Secondary parameters: Tmax, t½ and Kel were estimated in order to characterize rate and extent of absorption of the investigational drug products.
The sustained release tablets were very well absorbed and the extent of absorption was comparatively higher than that of the immediate release tablet. The sustained-release formulation was significantly lower than that of the marketed immediate-release. Tmax of sustained-release formulation was significantly long-lasting than that of immediate release. These outcomes are very much comparable with the profile of a sustained-release obvious by the lower elimination rate and higher t½ values. The bioavailability of sustained-release tablets remained the indistinguishable as that of immediate release tablets.
CONCLUSION
The test product and reference product are identical in terms of rate and extent of absorption. No major abnormalities or variations were reported due to test or reference medication in subject clinical and laboratory parameters. The overall conclusion was that both the medications at said dose are safe for administration.
ACKNOWLEDGMENT
The authors show gratefulness to the management for providing necessary support to carry out the work.
REFERENCES
- Silvati, Y., P. Karttunen, H. Tukiainen, P. Kokkonen, U. Hanninen and S. Nykanen, 1987. Pharmacokinetics of dextromethorphan and dextrorphan: A single dose comparison of three preparations in human volunteers. Int. J. Clin. Pharmacol. Ther. Toxicol., 25: 493-497.
PubMed - Woodworth, J.R., S.R. Dennis, O.N. Hinsvark, L.P. Amsel and K.S. Rotenberg, 1987. Bioavailability evaluation of a controlled-release dextromethorphan liquid. J. Clin. Pharmacol., 27: 133-138.
PubMed - Demirbas, S., L. Reyderman and S. Stavchansky, 1998. Bioavailability of dextromethorphan (as dextrorphan) from sustained release formulations in the presence of guaifenesin in human volunteers. Biopharm. Drug Dispos., 19: 541-545.
CrossRefDirect Link - Woodworth, J.R., S.R. Dennis, L. Moore and K.S. Rotenberg, 1987. The polymorphic metabolism of dextromethorphan. J. Clin. Pharmacol., 27: 139-143.
CrossRefDirect Link - Ramachander, G., F.D. Williams and J.F. Emele, 1977. Determination of dextrorphan in plasma and evaluation of bioavailability of dextromethorphan hydrobromide in humans. J. Pharm. Sci., 66: 1047-1048.
CrossRefDirect Link - Silvasti, M., P. Karttunen, P. Happonen, M. Mykkanen, T. Romppanen and H. Tukiainen, 1990. Pharmacokinetic comparison of a dextromethorphan-salbutamol combination tablet and a plain dextromethorphan tablet. Int. J. Clin. Pharmacol. Ther. Toxicol., 28: 268-272.
PubMedDirect Link - Yeh, G.C., P.L. Tao, H.O. Ho, Y.J. Lee, J.Y.R. Chen and M.T. Sheu, 2003. Analysis of pharmacokinetic parameters for assessment of dextromethorphan metabolic phenotypes. J. Biomed. Sci., 10: 552-564.
CrossRef - Kukanich, B. and M.G. Papich, 2004. Plasma profile and pharmacokinetics of Dextromethorphan after intravenous and oral administration in healthy dogs. J. Vet. Pharmacol. Ther., 27: 337-341.
CrossRef - Hu, L., L. Li, X. Yang, W. Liu and J. Yang et al., 2011. Floating matrix dosage form for Dextromethorphan hydrobromide based on gas forming technique: In vitro and in vivo evaluation in healthy volunteers. Eur. J. Pharm. Sci., 42: 99-105.
CrossRefDirect Link - Liu, D., X.Y. Chen, Y.F. Zhang, D.F. Zhong, Z. Gu and Y. Zhang, 2004. Determination of dextrorphan in human plasma and pharmacokinetic study. Acta Pharmaceutica Sinica, 39: 449-452.
Direct Link - Fossati, A., M.G. Vimercati, R. Caputo, L. Citerio, R. Ceriani and M. Valenti, 1993. Comparative pharmacokinetics of oral dextromethorphan and dextrorphan in the rabbit. Arzneimittel-Forschung, 43: 1337-1340.
Direct Link - Kazis, A., V. Kimiskidis and I. Niopas, 1996. Pharmacokinetics of dextromethorphan and dextrorphan in epileptic patients. Acta Neurol. Scand., 93: 94-98.
PubMed