
S. Rajamanikandan
Department of Bioinformatics, Alagappa University, Karaikudi-630 003, Tamil Nadu, India
R. Vanajothi
Department of Bioinformatics, Alagappa University, Karaikudi-630 003, Tamil Nadu, India
A. Sudha
Department of Bioinformatics, Alagappa University, Karaikudi-630 003, Tamil Nadu, India
P. Rameshthangam
Department of Biotechnology, Alagappa University, Karaikudi-630 003, Tamil Nadu, India
P. Srinivasan
Department of Bioinformatics, Alagappa University, Karaikudi-630 003, Tamil Nadu, India
Journal of Biological Sciences
Year: 2012 | Volume: 12 | Issue: 2 | Page No.: 83-90







ABSTRACT
Fibroblast growth factor receptor 2 (FGFR2) has important roles in embryonic development and tissue repair, especially bone and blood vessels. Many Single Nucleotide Polymorphism (SNPs) found in FGFR2 gene has been associated with various disorders such as crouzon, jackson-weiss syndromes, breast, ovarian and lung cancers. In this study, we performed a comprehensive analysis of the structural and functional impacts of all known SNPs in FGFR2 gene using publicly available computational prediction tools. Out of total 2255 SNPs retrieved from dbSNP, we found 58 non-synonymous SNP, 39 SNPs in the non-coding region which comprises 28 in 3’ UTR and 11 were found in 5’ UTR region. Among these SNPs, 10 non-synonymous SNPs were found to be damaging by both sequence homology based tool (SIFT) and structural homology based tool (PolyPhen). Untranslated region resource tools UTRscan and FastSNP were used to analyze the region which might change protein expression levels. Further, we modeled mutant protein and compared the energy minimization based on the native protein and identified major mutations from Tryptophan to Arginine and Tryptophan to Cysteine at position 290 of the native protein that caused the greatest impact on stability. From our results, we suggest two non-synonmous SNPs rs121918501 and rs121918499 could be a potential candidate for future studies on FGFR2 mutations.
PDF Abstract XML References Citation
Received: December 14, 2011;
Accepted: January 16, 2012;
Published: March 13, 2012
How to cite this article
S. Rajamanikandan, R. Vanajothi, A. Sudha, P. Rameshthangam and P. Srinivasan, 2012. In silico Analysis of Deleterious SNPs of the FGFR2 Gene. Journal of Biological Sciences, 12: 83-90.
DOI: 10.3923/jbs.2012.83.90
URL: https://scialert.net/abstract/?doi=jbs.2012.83.90
DOI: 10.3923/jbs.2012.83.90
URL: https://scialert.net/abstract/?doi=jbs.2012.83.90
INTRODUCTION
Fibroblast growth factors (FGFs) play important roles in the multiple cell biological activities, such as proliferation, differentiation, mitogenesis, migration and apoptosis and thus implicated in tumorigenesis (Dmowski et al., 2001; Eswarakumar et al., 2005; Taniguchi et al., 2000). Signaling is mediated through a family of four transmembrane receptor tyrosine kinases, a number of which are deregulated in development and neoplastic condition (Givol and Yayon, 1992; Goldfarb, 1996). Fibroblast growth factor receptor 2 (FGFR2) genes is composed of an extracellular region (consisting of three immunoglobulin like-domains), a transmembrane domain and a pair of intracellular tyrosine kinase domains. The second and third immunoglobulin like domains are involved in ligand binding. FGFR2 exons IIIa and IIIc together form the third immunoglobulin-like domain and are the most common sites for mutations involved in Crouzon syndrome (Meyers et al., 1996). In addition to Crouzon syndrome, mutations within the FGFR2 gene have also been identified in a number of familial and sporadic cases of Jackson-Weiss, Pfeiffer and Apert syndromes (Park et al., 1995; Malcolm and Reardon, 1996). As an indication of the complex interactions that govern genotype-phenotype outcomes, an identical mutation in exon IIIc of the FGFR2 gene is shared by Crouzon and Jackson-Weiss syndromes (Gorry et al., 1995).
Single Nucleotide Polymorphisms (SNPs) play a major role in the understanding of the genetic basis of many complex human diseases. SNPs are mostly bi-allelic polymorphism and may occur in both coding and non-coding region of the genome. SNPs found within a coding region are of particular interest of scientist as they play major role in the alteration of biological function of protein (Javed and Mukesh, 2010). About 500,000 SNPs fall into the coding regions of the human genome (Collins et al., 1998). Many SNPs have no effect on cell function; however, others could predispose people to disease or influence their response to a drug. The occurrence of a SNP in the genome is at random and they are reported to occur in a given population consistently. Hence, these are used as ideal biomarkers (Khan and Jamil, 2008). SNPs of FGFR2 gene has showed the increased risk of breast cancer (Hunter et al., 2007) and its association was confirmed by using candidate-gene approach (Huijts et al., 2007). Mutation in FGFR2 gene occurs in variety of cancer such as gastric, lung, ovarian and endometrial uterus and this suggests that these receptor tyrosine kinases have an overlapping function in structure biology.
Non-synonymous SNPs (nsSNPs) that lead to an amino acid residue substitution in the protein products are of particular interest because they are responsible for nearly half of the known genetic variation related to human inherited disease (Krawczak et al., 2000). These are likely to be an important factor contributing to the functional diversity of the encoded proteins in the human population (Lander, 1996). Discovering the deleterious nsSNPs is the important task in the pharmacogenomics to evaluate drug response experimentally. From this view, we carried out the computational analysis of the nsSNPs in the FGFR2 gene to identify possible deleterious mutations. Based on the results, we proposed a modeled structure for the mutant proteins to check its stability.
MATERIALS AND METHODS
Datasets: Database of Single Nucleotide Polymorphism (dbSNP) (http://www.ncbi.nlm.nih.gov/SNP) were used to retrieve SNPs and their related protein sequences in FGFR2 gene for systematic approach.
Analysis of functional consequences of coding nsSNPs by SIFT method: SIFT (Ng and Henikoff, 2003) is a sequence homology based tool that sorts intolerant from tolerant amino acid substitutions and predicts whether an amino acid substitution at a particular position in a protein will have a phenotypic effect. It also predicts the conservation level of a particular position in a protein. SIFT takes a query sequence and uses multiple alignment information to predict tolerated and deleterious substitutions for every position of the query sequence. SIFT is a multistep procedure that, given a protein sequence (1) searches for similar sequences (2) chooses closely related sequences that may share similar function (3) obtains the multiple alignment of these chosen sequences and (4) calculated normalized probabilities for all possible substitutions at each position from the alignment. Substitutions at each position with normalized probabilities less than a chosen cutoff are predicted to be deleterious; those greater than or equal to the cutoff are predicted to be tolerated. The cutoff values ranges from 0.00 to 0.05 was predicted to be intolerance/deleterious. The higher the tolerance index (above 0.05); will have less functional impact of the particular amino acid substitution.
Simulation for functional change in coding nsSNPs by PolyPhen: PolyPhen (Polymorphism phenotyping) (Ramensky et al., 2002) were used to predict possible impact of amino acid substitution on the structure and function of human protein(s). Analyzing the damaged coding non-synonymous SNPs at the structural level is considered to be important factor for understanding the functional activity of the protein. PolyPhen server gets input either as amino acid sequence of a protein or the SWALL database ID or accession number together with sequence position and two amino acid variants characterizing the polymorphism. Sequence based homologous sequences and mapping of substitution site to a know protein’s 3-Dimensional structure are the parameters taken into account by the PolyPhen server. It calculates Position Specific Independent Counts (PSIC) scores for each of the two variants and then computes the difference between the PSIC scores. The higher the PSIC score difference will have higher the functional impact for the particular amino acid substitution.
Functional significance of noncoding SNPs in regulatory untranslated regions: FastSNP (function analysis and selection tool for single nucleotide polymorphism) were used to identify the SNPs that are most likely to have functional effects (Yuan et al., 2006). 5’ UTR regions were predicted by FastSNP server. Recent studies show that SNPs may have functional effects on protein structures, by changing single amino acids, transcriptional regulation and on alternative splicing regulations (Prokunina and Alarcon-Riquelme, 2004; Prokunina et al., 2002). The FastSNP server follows the decision tree principle with external web service access to TF search, to obtain the predicted transcription factor-binding sites of the gene. Thus the decision tree will assign risk rankings for SNP prioritization with a ranking of 0, 1, 2, 3, 4 and 5. This signifies the levels of no, very low, medium, high and very high effects, respectively.
Scanning of UTR SNPs in UTR site: The 5’ and 3’ UTR regions are involved in various biological processes such as post-transcriptional regulatory pathways stability and translational efficiency (Sonenberg, 1994; Nowak, 1994). We used the program UTRscan (Pesole and Liuni, 1999) server to predict 3’ UTR region alone. This allows the search of user-submitted sequences for any patterns collected in the UTR site. UTR site is a collection of functional sequence patterns located in 5 and 3 UTR sequences. UTRscan which looks for UTR functional elements by searching through user-submitted sequence data for the patterns defined in the UTR site and UTR database. If the different UTR SNP is found to have different functional pattern(s) the UTR SNP is predicted to have functional significance. The internet resources for UTR analysis are UTRdb and UTR site. UTRdb contains experimentally proven biological activity of functional sequence patterns of UTR sequences from eukaryotic mRNAs (Pesole et al., 2002).
Modeling of mutant structure and calculation of their RMSD difference: Structural analysis was performed in order to evaluate and compare the stability of native and mutant structures. The native structure of FGFR2 protein was available in the Protein Data Bank (PDB ID: 1djs) with highest resolution (2.40 Å) (Stauber et al., 2000). We confirmed the mutation positions and the mutation residues using the dbSNP database (Sherry et al., 2001). The mutation and energy minimization for the 3D structure was performed by using the SWISSPDB viewer. Divergence of mutant structure from native structure is due to mutation, deletions and insertions (Rajasekaran et al., 2008) and the deviation between the two structures is evaluated by their RMSD values which could affect stability and functional activity (Rasricha et al., 2011).
Computational analysis of stabilizing residues: We used the server SRide (Magyar et al., 2005) for identifying the stabilizing residues in native protein and in the mutant model to check their stability. The input option for this server is the atomic coordinate file of the protein to be analyzed. Alternatively we can upload the PDB format directly; this option is mainly to analyze structures obtained by homology modeling or other computational approaches. Stabilizing residues were computed using parameters such as surrounding hydrophobicity, long-range order, stabilization center and conservation score.
RESULTS AND DISCUSSION
SNP dataset: SNPs have become marker of choice for many applications in genome analysis because SNPs are abundance, stable, ubiquity and interspersed in nuclear genome (Tchin et al., 2011). Single nucleotide polymorphism data of FGFR2 gene was retrieved from the dbSNP database. It contained a total of 2255 SNPs, of which 58 were non-synonymous, 39 were in non-coding region which comprises 11 SNPs in 5’ UTR, 28 SNPs in 3’ UTR and the rest SNPs were found in the intronic region. We took non-synonymous, 5’UTR and 3’UTR SNPs for our computational analysis. The distribution of these SNPs in coding and noncoding regions was illustrated in Fig. 1.
Deleterious nsSNPs by SIFT program: The conservation level of the particular position of the protein was determined using the sequence homology based method. The protein accession number with mutational position with two amino acid variants associated to the 58 nsSNPs were submitted as input to the SIFT server to check its tolerance index.
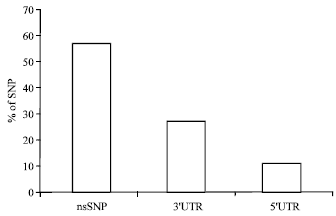 | |
| Fig. 1: | Distribution of nonsynonymous, 5’ UTR and 3’ UTR SNPs |
| Table 1: | List of nsSNPs that were predicted to have functional significance by SIFT |
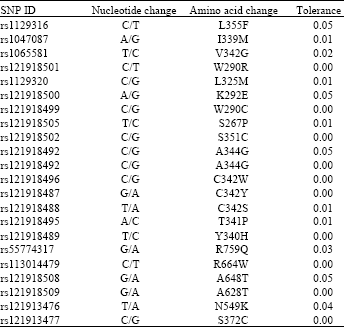 | |
Lower the tolerance index, higher will be the functional impact of the amino acid substitution (Ng and Henikoff, 2001). Among the 58 nsSNPs, 22 were found to be deleterious, having a tolerance index score of p = 0.05. The results are summarized in Table 1. We observed that of 58 deleterious nsSNPs, 10 showed a high deleterious tolerance index score of 0.00. The remaining 5 nsSNPs showed a tolerance index score of 0.01, followed by 1, 1, 1 and 4 nsSNPs with a tolerance index of 0.02, 0.03, 0.04 and 0.05, respectively. On analyzing the nucleotide change, seven nsSNPs showed a nucleotide change of C/G, four a change from G/A, three a change from C/T and T/C, two a change from A/G and T/A and one a change from A/C. The nucleotide changes from C/G occurred a maximum number of times which accounts for the highest number of deleterious nsSNPs with a SIFT tolerance index of 0.00.
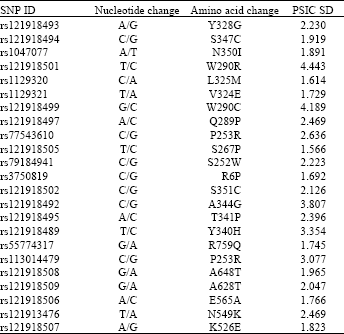
Damaged nsSNP found by the PolyPhen server: The structural level of the protein was analyzed using the PolyPhen server. All the 58 protein sequences of non-synonmous SNPs submitted to SIFT were also submitted to the PolyPhen server. A Position-Specific Independent Count (PSIC) score difference of 1.5 and above is considered to be damaging (Ramensky et al., 2002). Out of 58 nsSNPs, 23 nsSNPs were considered to be damaging and exhibited a PSIC score difference between 1.566 and 4.443 was showed in Table 2. Ten nsSNPs that was observed to be deleterious by the SIFT program was also found to be damaging according to PolyPhen. Hence, we could infer that the results obtained from SIFT shows a significant correlation with the results obtained from the PolyPhen which specifies the structural details. From Table 1 and 2, it was clear that the non-synonmous SNPs (rs121918501 and rs121918499) may disrupt both the structure and function of the protein coded by FGFR2 gene and it could be précised important for the recognition of various disorders such as Crouzon and Jackson-Weiss syndromes, breast, ovarian and lung cancers. The most damaging non-synonmous SNPs (rs121918501 and rs121918499) showed a PSIC score of 4.443 and 4.189 with a SIFT tolerance index of 0.00, due to the mutation from Tryptophan to Arginine and Tryptophan to Cysteine. Meanwhile, other studies had also reported on the association of SNPs with various applications such as the search for SNPs in PS1 in AD patients (Al-Khedhairy et al., 2005), identification of polymorphic sites (1236 and 3435) in multi drug resistance gene 1 Influencing drug response in breast cancer patients (Khan et al., 2007), genetic polymorphisms in the ESR genes and the risk of breast cancer among Iranian women (Abbasi et al., 2009a), genome wide SNP analysis in Mycobacterium sp. (Srivastava et al., 2009).
| Table 2: | List of nsSNPs that were predicted to have functional significance by PolyPhen |
 | |
Functional SNPs in UTR found by the FastSNP: We used FastSNP server to analyze the SNPs in 3’ and 5’ untranslated region that are predicted to be functionally significant (Yuan et al., 2006). According to this server, of 11 5’ UTR SNPs present in the FGFR2 gene, two 5’ UTR namely rs3135721 and rs41258305 were predicted to be damaging with a risk ranking of 1-3 (Table 3). The nucleotide changes were G/T for SNP ID rs3135721 and C/T for SNP ID rs41258305. 3’UTR SNPs were not predicted by this server.
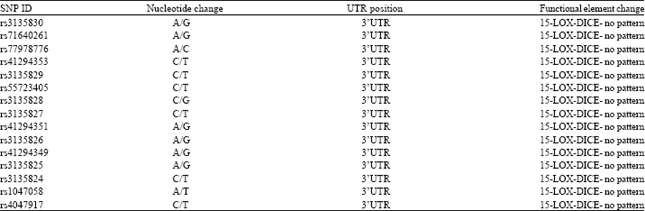
Functional SNPs in UTR found by the UTRscan server: Polymorphisms in the 3’ UTR affect gene expression by affecting the ribosomal translation of mRNA or by influencing the RNA half-life (Deventer, 2000). We analyzed 28 SNPs in the 3’ untranslated region using the UTRscan server. The UTRscan server finds patterns of regulatory region motifs from the UTR database and gives information about whether the matched pattern is damaged. About fifteen UTR SNPs were predicted to be damaging by this server and presented in Table 4. The 15-lipoxygenase (15-LOX) differentiation control element (15-LOX-DICE) controls 15-LOX synthesis which catalyzes the degradation of lipids and is an important factor responsible for the degradation of mitochondria during reticulocyte maturation (Rajasekaran et al., 2007). This 15-LOX-DICE exists in the fifteen 3’ UTR SNPs that were considered to be of functional significance and it may lead to FGFR2 gene damage. No reports have been analysed so far relating the deleterious nature of SNPs with untranslated regions of FGFR2 gene.
Modeling of mutant structure and calculation of their RMSD difference: Single amino acid mutations can significantly change the stability of a protein structure. So the knowledge of a protein’s three-dimensional (3D) structure is essential for understanding of its functionality (Chen and Shen, 2009). Information about mapping the deleterious nsSNPs in the protein structure was obtained from dbSNP. The available structure for the FGFR2 gene has the PDB ID 1djs (Fig. 2). Two nsSNPs (rs121918501 and rs121918499) were selected for structural analysis. The mutational position and amino acid variant associated with these nsSNPs is W/R at the residue position 290 and W/C at the residue position 290 was mapped in the 1djs native structure.
| Table 3: | List of SNPs (UTR mRNA) predicted to be functionally significant by FastSNP |
| Table 4: | List of SNPs (UTR mRNA) predicted to be functionally significant by UTRscan |
 | |
| Table 5: | Stabilizing residues in native and mutant models of 1djs by SRide |
 | |
| Fig. 2: | Native structure of FGFR2 gene (1djs) |
Mutation and energy minimization at the specified position was performed by SWISSPDB viewer to obtain a modeled structure for both the native structure (1djs) and mutant modeled structures (W290R) and (W290C). It can be seen that the total energy for the mutant modeled structures were found to be -15249.33 and -14932.46 Kcal moL-1, respectively. The super impositions between the native and mutant structures were performed using Pymol suite and RMSD values was found to be 0.40Å and 0.45Å, respectively. Since these values are low, we can suggest that these mutations do not cause a significant change in the mutant structures with respect to native protein structure. Mutation and polymorphism of cancer-associated genes have been found to predict tumor formation and prognosis. It is also considered as an effective risk factor with positive effects and negative effects in different studies (Abbasi et al., 2009b). The superimposed structures of the native protein 1djs and the two mutant type proteins are shown in Fig. 3a and b, respectively.
Computing stabilizing residues between native structure and mutant modeled structures: We used the SRide server to identify the stabilizing residues of the native-type structure and mutant modeled structures. The results are summarized in Table 5. Six residues were identified in the native-type 1djs structure. Four stabilizing residues were identified in mutant model 1djs (W290R) and seven were identified in mutant model 1djs (W290C). Three stabilizing residues, namely Ala (181), Val (248) and Cys (342) were found to be common to both the native structure 1djs and mutant models. The remaining residues were not found common in mutant and native structures. Higher number of stabilizing residues in the mutant type 1djs (W290C) matched with the native protein structure compared to the mutant type (W290R). Therefore we predict that the mutation from Tryptophan to Cysteine in the native type protein will be more deleterious than Tryptophan to Arginine and hence both SNPs could be important candidates for various disorders in the FGFR2 gene.
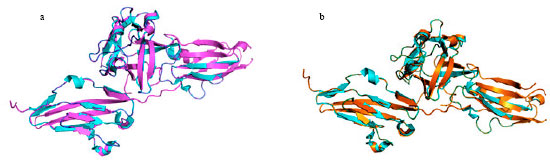 | |
| Fig. 3(a-b): | (a) Superimposed structure of native protein 1djs (cyan color) with mutant protein W290R magenta color (b) Superimposed structure of native protein 1djs (cyan color) with mutant protein W290C orange color |
CONCLUSION
In the present study, we investigated the functional and structural impact of SNPs in the FGFR2 gene by computational prediction tools. Out of a total 2255 SNPs in the FGFR2 gene, we found 58 non-synonmous SNPs, 28 3’ UTR and 11 were found in 5’ UTR region and the remaining 2158 were found in intronic region. Out of 58 nsSNPs, 10 nsSNPs were found to be deleterious by both SIFT and PolyPhen server. Two SNPs in the 5’ UTR and 15 SNPs in the 3’ UTR were found to be of functional significance. It was found that the major mutation in the native protein of the FGFR2 gene was from Tryptophan to Arginine and Tryptophan to Cysteine. Of two nsSNPs that had this mutation, we conclude that rs121918501 with a mutation of Tryptophan to Arginine and rs121918499 with a mutation of Tryptophan to Cysteine at position 290 in the native protein 1djs could be the main target for the diseases caused by the FGFR2 gene.
ACKNOWLEDGMENT
The authors gratefully acknowledge the computational and bioinformatics facility provided by the Department of Bioinformatics, Alagappa University, Karaikudi-630003, Tamilnadu, India.
REFERENCES
- Abbasi, S., C. Azimi, F. Othman, M.R. Noori Daloii and Z.O. Ashtiani et al., 2009. Estrogen receptor-α gene codon 10 (T392C) polymorphism in Iranian women with breast cancer: A case study. Trends Mol. Sci., 1: 1-10.
CrossRefDirect Link - Abbasi, S., P. Ismail., F. Othman., R. Rosli and C. Azimi, 2009. Estrogen receptor-α gene, codon 594 (G3242A) polymorphism among iranian women with breast cancer: A case control study. Asian J. Sci. Res., 2: 51-60.
CrossRefDirect Link - Al-Khedhairy, A.A.A., M. Arfin , B.A. Alahmadi and M.A. Al-Jumah, 2005. Single nucleotide polymorphism associated with late-onset Alzheimer's disease. J. Medical Sci., 5: 275-279.
CrossRefDirect Link - Chen, J. and B. Shen, 2009. Computational analysis of amino acid mutation: A proteome wide perspective. Curr. Proteomics., 63: 228-234.
Direct Link - Collins, F.S., L.D. Brooks and A. Chakravarti, 1998. A DNA polymorphism discovery resource for research on human genetic variations. Genome Res., 8: 1229-1231.
PubMed - Deventer, S.J.V., 2000. Cytokine and cytokine receptor polymorphisms in infectious disease. Intensive Care Med., 26: S98-S102.
PubMedDirect Link - Dmowski, W.P., J. Ding, J. Shen, N. Rana, B.B. Fernandez and D.P. Braun, 2001. Apoptosis in endometrial glandular and stromal cells in women with and without endometriosis. Hum. Reprod., 16: 1802-1808.
CrossRefDirect Link - Eswarakumar, V.P., I. Lax and J. Schlessinger, 2005. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev., 16: 139-149.
CrossRefPubMedDirect Link - Givol, D. and A. Yayon, 1992. Complexity of FGF receptors: Genetic basis for structural diversity and functional specificity. Federat. Am. Soc. Exp. Biol., 64: 3362-3369.
Direct Link - Goldfarb, M., 1996. Functions of fibroblast growth factors in vertebrate development. Cytokine Growth Factor Rev., 7: 311-325.
PubMedDirect Link - Gorry, M.C., R.A. Preston, G.J. White,Y. Zhang and V.K. Singhal et al., 1995. Crouzon syndrome: Mutations in two spliceoforms of FGFR2 and a common point mutation shared with Jackson-Weiss syndrome. Hum. Mol. Genet., 42: 1387-1390.
PubMedDirect Link - Huijts, P.E.A, M.P.G. Vreeswijk, K.H.G. Kroeze-Jansema, C.E. Jacobi and C. Seynaeve et al., 2007. Clinical correlates of low-risk variants in FGFR2, TNRC9, MAP3K1, LSP1 and 8q24 in Dutch cohort of incident breast cancer case. Breast Cancer Res., Vol. 9.
CrossRefDirect Link - Hunter, D.J., P. Kraft, K.B. Jacobs, D.G. Cox and M. Yeager et al., 2007. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat. Genet., 39: 870-874.
CrossRefDirect Link - Javed, R. and Mukesh, 2010. Current research status, databases and application of single nucleotide polymorphism. Pak. J. Biol. Sci., 13: 657-663.
CrossRefDirect Link - Khan, S., K. Jamil, G.P. Das , C.M. Vamsy and S. Murthy, 2007. Polymorphic sites (1236 and 3435) in multi drug resistance gene 1 influencing drug response in breast cancer patients. Int. J. Pharmacol., 3: 453-460.
CrossRefDirect Link - Khan, M. and K. Jamil, 2008. Study on the conserved and the polymorphic sites of MTHFR using bioinformatics approaches. Trends Bioinform., 1: 7-17.
CrossRefDirect Link - Krawczak, M., E.V. Ball and I. Fenton, 2000. Human gene mutation database: A biomedical information and research resource. Hum. Mutat., 15: 45-51.
PubMedDirect Link - Magyar, C., M.M. Gromiha, G. Pujadas, G.E. Tusnady, I. Tusnady and S. Simon, 2005. SRide: A server for identifying stabilizing residues in proteins. Nucleic Acids Res., 33: W303-W305.
PubMedDirect Link - Meyers, G.A., D. Day, R. Goldberg, D.L. Daentl and K.A. Przylepa et al., 1996. FGFR2 exon IIIa and IIIc mutations in Crouzon, Jackson-Weiss, and Pfeiffer syndromes: evidence for missense changes, insertions, and a deletion due to alternative RNA splicing. Am. J. Hum. Genet., 58: 491-498.
PubMed - Ng, C.P. and S. Henikoff, 2001. Predicting deleterious amino acid substitutions. Genome Res., 11: 863-874.
PubMed - Ng, P.C. and S. Henikoff, 2003. SIFT: Predicting amino acid changes that affect protein function. Nucl. Acids Res., 31: 3812-3814.
CrossRefDirect Link - Park, W.J., G.A. Bellus and E.W. Jabs, 1995. Mutations in fibroblast growth factor receptors: phenotypic consequences during eukaryotic development. Am. J. Hum. Genet., 57: 748-754.
PubMed - Pesole, G., S. Liuni, G. Grillo, F. Licciulli, F. Mignone, C. Gissi and C. Saccone, 2002. UTRdb and UTRsite: Specialized databases of sequences and functional elements of 5' and 3' untranslated regions of eukaryotic mRNAs. Nucleic Acids Res., 30: 335-340.
PubMed - Prokunina, L., C. Castillejo-Lopez, F. Oberg, I. Gunnarsson and L. Berg et al., 2002. A regulatory polymorphism in PDCD1 is associated with susceptibility to systemic lupus erythematosus in humans. Nat. Genet., 32: 666-669.
PubMed - Prokunina, L. and M.E. Alarcon-Riquelme, 2004. Regulatory SNPs in complex diseases: Their identification and functional validation. Expert Rev. Mol. Med., 6: 1-15.
PubMed - Rajasekaran, R., C. Sundandiradoss., C.G. Doss and R. Sethumadhavan, 2007. Identification and in silico analysis of functional SNPs of the BRCA1 gene. Genomics, 90: 447-452.
PubMed - Rajasekaran, R., G.P. Doss, C. Sudandiradoss, K. Ramanathan, P. Rituraj and R. Sethumadhavan, 2008. Computational and structural investigation of deleterious functional SNPs in breast cancer BRCA2 gene. Sheng Wu Gong Cheng Xue Bao, 24: 851-856.
PubMed - Ramensky, V., P. Bork and S. Sunyaev, 2002. Human non-synonymous SNPs: Server and survey. Nucl. Acids Res., 30: 3894-3900.
PubMed - Rasricha, R., A. Chandolia, P. Ponnan, N.K. Sainim and S. Sharma et al., 2011. Single nucleotide polymorphism in the genes of mce1 and mce4 operons of Mycobacterium tuberculosis: analysis of clinical isolates and standard reference strains. BMC Microbiol., 11: 41-41.
PubMed - Sherry, S., M. Ward, M. Kholodov, J. Baker, L. Phan, E.M. Smigielski and K. Sirotkin, 2001. dbSNP: The NCBI database of genetic variation. Nucl. Acids Res., 29: 308-311.
PubMed - Srivastava, S.K., M. Agrawal and M. Grover, 2009. Genome wide single nucleotide polymorphism analysis of mycobacterium species and subspecies. Res. J. Microbiol., 4: 112-121.
CrossRefDirect Link - Sonenberg, N., 1994. mRNA translation: influence of the 5′ and 3′ untranslated regions. Curr. Opin. Genet. Dev., 4: 310-315.
PubMed - Stauber, D.J., A.D. DiGabriele and W.A. Hendrickson, 2000. Structural interactions of fibroblast growth factor receptor with its ligands. Proc. Natl. Acad. Sci., 97: 49-54.
CrossRefDirect Link - Taniguchi, F., T. Harada, M. Ito, S. Yoshida, T. Iwabe, M. Tanikawa and N. Terakawa, 2000. Keratinocyte growth factor in the promotion of human chorionic gonadotropin production in human choriocarcinoma cells. Am. J. Obstet. Gynecol., 182: 692-698.
PubMedDirect Link - Tchin, B.L., W.S. Ho, S.L. Pang and J. Ismail, 2011. Gene-associated single nucleotide polymorphism (SNP) in Cinnamate 4-Hydroxylase (C4H) and Cinnamyl alcohol dehydrogenase (CAD) genes from Acacia mangium superbulk trees. Biotechnology, 10: 303-315.
CrossRef - Yuan, H.Y., J.J. Chiou, W.H. Tseng, C.H. Liu and C.K. Liu et al., 2006. FASTSNP: An always up-to-date and extendable service for SNP function analysis and prioritization. Nucl. Acids Res., 34: W635-W641.
PubMed