
J.A. Ansari
Department of Pharmacology, Faculty of Pharmacy, Hamdard University, New Delhi-110062, India
Journal of Biological Sciences
Year: 2010 | Volume: 10 | Issue: 5 | Page No.: 386-395







ABSTRACT
Drugs are an important reason of hepatotoxicity. In general more than 900 drugs, toxins and herbs have been reported to cause hepatotoxicity and drugs account for 20-40% of all instances of fulminant hepatic failure. Specific therapy against drug-induced hepatotoxicity is limited to the use of N-acetylcysteine in the early phases of paracetamol toxicity. L-carnitine is potentially valuable in cases of valproate toxicity. In general, corticosteroids have no definitive role in treatment. They may prevent the systemic features associated with hypersensitivity or allergic reactions. Cholestyramine can be used for alleviation of pruritus. Ursodeoxycholic acid may be used. Lastly, consulting a hepatologist is always useful for other agents, supportive measures and the increasing use of liver-assist devices as well as emergency liver transplantation are available when drug injury evolves into irreversible liver failure. It is expected that a better understanding of hepatotoxicity mechanisms will lead to the development of more specific and effective forms of therapy in the near future.
PDF Abstract XML References Citation
Received: March 19, 2010;
Accepted: May 28, 2010;
Published: July 14, 2010
How to cite this article
J.A. Ansari, 2010. Therapeutic Approaches in Management of Drug-induced Hepatotoxicity. Journal of Biological Sciences, 10: 386-395.
DOI: 10.3923/jbs.2010.386.395
URL: https://scialert.net/abstract/?doi=jbs.2010.386.395
DOI: 10.3923/jbs.2010.386.395
URL: https://scialert.net/abstract/?doi=jbs.2010.386.395
INTRODUCTION
Drugs are an important reason of hepatotoxicity. In general more than 900 drugs, toxins and herbs have been reported to cause hepatotoxicity and drugs account for 20-40% of all instances of fulminant hepatic failure (Nilesh et al., 2009) (Table 1). Acute hepatitis, with or without cholestasis, is the usual histological pattern of DILI (drug-induced liver disease) and drugs such as acetaminophen are the leading causes of acute liver failure. Most cases of DILI resolve on discontinuation of the drug, but recovery can take months or rarely the disease can progress despite drug withdrawal. Drugs such as methotrexate may lead to chronic hepatitis and cirrhosis, while others such as minocycline, nitrofurantoin and methyldopa are implicated in autoimmune hepatitis.
| Table 1: | Drugs that have caused acute fulminant hepatic failure (Medical Economics, 2000) |
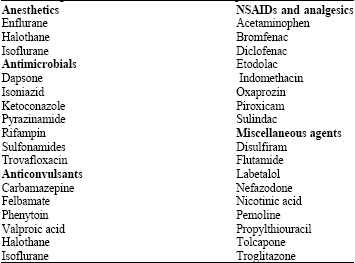 | |
Drug-induced steatohepatitis is not a common pattern, but is well described with drugs such as amiodarone and irinotecan. In the presence of risk factors such as obesity and diabetes, some drugs such as tamoxifen, oestrogens and nifedipine can cause or exacerbate steatohepatitis. Other observed patterns include granulomatous hepatitis, vascular injury (e.g., sinusoidal obstruction syndrome), cell lipidosis and neoplasms (e.g., adenomas) (Ramachandran and Kakar, 2009).
GENERAL CONSIDERATIONS
DILD is a potential complication of any prescribed medication, because of the central role of the liver in drug metabolism and elimination (Javed et al., 2009). Drug-induced hepatotoxic reactions are of several types and origin and the time to onset varies from being very short to exhibiting a long latency. Clinically, the most relevant reactions include liver necrosis, hepatitis, cholestasis, vascular changes and steatosis. It can be emphasized that species differences in drug metabolism, target molecule and pathobiology must be taken into account in the interpretation of findings and in assessing the relevance of such findings to humans. For example, steatosis has significant implications to clinicians, i.e., Non-Alcoholic Steato Hepatitis (NASH), yet it is generally a less important finding non-clinically, particularly if observed in rodents.
A drug can induce liver toxicity via several mechanisms. For example, it can be directly acting or indirectly through reactive metabolites. The drug or its metabolites may precipitate liver toxicity after specific receptor binding, or reactive metabolites can react with hepatic macromolecules leading to direct cytotoxicity. On the other hand, liver toxicity can be mediated via an immunological cascade. Increases in the levels of the liver enzymes alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in serum, in combination with increased bilirubin levels, are actually considered to be the most relevant indication of liver toxicity (CHMP, 2008).
CLINICAL IMPLICATIONS
Clinical pattern of hepatotoxicity: Generaly, hepatotoxicity caused by drugs is known to be either Type A dose dependent (intrinsic toxicity) or Type B idiosyncratic (Rawlins and Thompson, 1977; Park et al., 1998). Perhaps with the main exception of single high dose of acetaminophen-associated hepatotoxicity, most drug-induced hepatotoxicity cases, evaluated in clinical practice, are considered as idiosyncratic (Gunawan and Kaplowitz, 2004). Normally, predictable reactions can be detected at the preclinical and clinical stage of drug development. In general, these reactions are dose related (intentional or accidental). Predictable reactions have a short latency period, usually several hours to a few days (e.g., acetaminophen or chemotherapy drugs) (Walgren et al., 2005).
The mechanism behind hepatotoxicity is poorly understood. It can be accompanied with (1) immunoallergic features such as eosinophilia, rash, antibody titer and fever having variable, usually short latency period (1-6 weeks) or (2) proceed without immunoallergic manifestations and delayed latency period (up to 1 year) (Larrey, 2002; Egger et al., 2005). However, the absence of the common features of hypersensitivity does not exclude an immune mediated toxicity. These features are only present in 23% of the patients with drug induced hepatotoxicity (Andrade et al., 2005a). Many independent co-stimulatory factors may determine idiosyncratic drug induced hepatotoxicity such as environment, age, sex, infections and pharmacogenetic variation in drug metabolising polymorphisms between individuals (Table 2).
Important and specific agents with their effects on the liver.
Most drugs have a signature effect, which is a specific pattern of liver injury, although some drugs such as rifampin can cause all kinds of liver injury, including hepatocellular injury, cholestasis, or even isolated hyper bilirubinemia. However, knowledge of the most commonly implicated agents and a high index of suspicion are very essential in diagnosis (Nilesh et al., 2009).
| Table 2: | Overview of drug-induced hepatotoxicity patterns (Temple, 2002, Gunawan and Kaplowitz, 2004; Ibanez et al., 2002) |
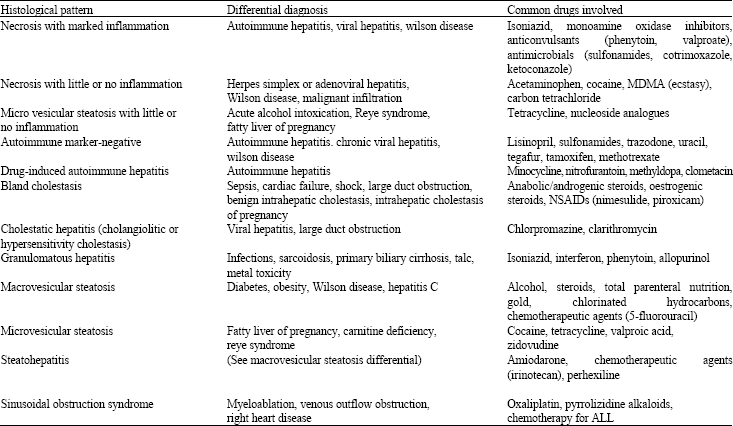 | |
| ALL: Acute lymphoblastic leukaemia; MDMA: 3,4-methylenedioxymethylamphetamine; NSAID: Non-steroidal anti-inflammatory drug | |
Paracetamol: Acetaminophen [N-acetyl-p-aminophenol (APAP)], either singly or as a component of a drug cocktail, is a frequently used over-the-counter and prescription medication worldwide for analgesic and antipyretic effects and it has a reasonable safety profile when consumed in therapeutic doses. In contrast, supratherapeutic doses (intentionally or inadvertently ingested) were recognized to cause severe liver toxicity and even fatal hepatic failure as early as the 1960s. APAP can also induce renal failure and eventually death in severe cases (Nelson, 1990).
The therapeutic serum concentration of APAP ranges from 10 to 20 μg mL-1. However, the diagnosis of APAP-induced hepatotoxicity should be entertained in a patient with a history of chronic, excessive APAP ingestion and elevated liver enzymes, regardless of measured serum levels. Toxicity is unlikely to occur with a single dose of less than 150 mg kg-1 in children or less than 7.5 g in adults. Conversely, it is likely to occur when amounts greater than 250 mg kg-1 in children or more than 12 g in adults are ingested over a 24 h period (Tan et al., 2009).
Amoxicillin: Amoxicillin precipitates a moderate rise in SGOT levels, SGPT levels, or both, but the significance of this finding is unknown. Hepatic dysfunction, including jaundice, hepatic cholestasis and acute cytolytic hepatitis, have been reported (Nilesh et al., 2009).
Amiodarone: Amiodarone causes abnormal liver function test results in 15-50% of patients. The spectrum of liver injury is wide, ranging from isolated asymptomatic transaminase elevations to a fulminant disorder. Hepatic dysfunction usually develops more than 1 year after starting therapy, but it can occur in 1 month. It is usually predictable, dose dependent and has a direct hepatotoxic effect. Some patients with elevated aminotransferase levels have detectable hepatomegaly and clinically important hepatic disease develops in less than 5% of patients. In rare cases, amiodarone toxicity manifests as alcoholic liver disease. Hepatic granulomas are rare. Importantly, amiodarone has a very long half-life and therefore may be present in the liver for several months after withdrawal of therapy. Amiodarone is iodinated and this result in increased density on CT scans, which does not correlate with hepatic injury (Chang et al., 1999; Morelli et al., 1991).
Aspirin: Aspirin is rapidly converted to salicylic acid after absorption. The major metabolites of aspirin are salicyluric and salicylphenolic glucuronide. Aspirin overdose leads to systemic toxicity, but liver failure is rare. The exact mechanism of the cellular injury is not clear, although several possible modes of action have been postulated. These include lipid peroxidation, mitochondrial damage, hydroxyl radical scavenging and toxicity to hepatocyte membranes (Zimmerman, 1981).
Chlorpromazine: Chlorpromazine liver toxicity resembles that of infectious hepatitis with laboratory features of obstructive jaundice rather than those of parenchymal damage. The overall incidence of jaundice is low irrespective of dose or indication of the drug. Most cases occur 2-4 weeks after therapy. Any surgical intervention should be withheld until extra hepatic obstruction is confirmed. It is usually promptly reversible upon withdrawal of the medication; however chronic jaundice has been reported. Chlorpromazine should be administered with caution to persons with liver dysfunction (Bass, 2003).
Ciprofloxacin: Cholestatic jaundice has been reported with repeated administration of quinolones. Approximately 1.9% of patients taking ciprofloxacin show elevated SGPT levels, 1.7% has elevated SGOT levels, 0.8% has increased alkaline phosphatase levels and 0.3% has elevated bilirubin levels. Jaundice is transient and enzyme levels return to the reference range (Bass, 2003).
Diclofenac: Elderly females are more susceptible to diclofenac-induced liver injury. Elevations of one or more liver test results may occur. These laboratory abnormalities may progress, may remain unchanged, or may be transient with regular therapy. Borderline or greater elevations of transaminase levels occur in approximately 15% of patients treated with diclofenac. Of the hepatic enzymes, ALT is recommended for monitoring liver injury. Meaningful (>3 times the upper limit of the reference range) elevations of ALT or AST occur in approximately 2% of patients during the first 2 months of treatment. In patients receiving long-term therapy, transaminase levels should be measured periodically within 4-8 weeks of initiating treatment (Batt and Ferrari, 1995).
Erythromycin: Erythromycin may cause hepatic disorder, including increased liver enzyme levels and hepatocellular and/or cholestatic hepatitis with or without jaundice. A cholestatic reaction is the most common adverse effect and usually begins within 2-3 weeks of therapy. The liver mainly excretes erythromycin; exercise caution when this drug is administered to patients with impaired liver function. The use of erythromycin in patients concurrently taking drugs metabolized by the P-450 system may be associated with elevations in the serum levels of other drugs (Bass, 2003).
Ethambutol: There are fewer reports of hepatic dysfunction with ethambutol in the treatment of tuberculosis. Abnormal liver function tests have been reported in some patients taking ethambutol; however, these patients were also taking other anti tubercular drugs known to cause liver dysfunction (Tahaoglu et al., 2001).
Fluconazole: The spectrum of hepatic reactions ranges from mild transient elevations in transaminase levels to hepatitis, cholestasis and fulminant hepatic failure. In fluconazole-associated liver toxicity, hepatotoxicity is not obviously related to the total daily dose, duration of therapy, or sex or age of the patient. Fatal reactions occur in patients with serious underlying medical manifestations. Fluconazole-associated hepatotoxicity is usually, but not always, reversible upon discontinuation of therapy (Bass, 2003).
Isoniazid: Around 10-20% of patients during the first 4-6 months of therapy have a mild hepatic dysfunction shown by mild and transient increase in serum AST, ALT and bilirubin concentration. But in some patients the hepatic damage can be progressive and cause fatal hepatitis. Acetyl hydrazine, a metabolite of Isoniazid is responsible for liver damage. Isoniazid should be stopped if the AST increases to over 5 times the normal value. A prospective cohort study of 11,141 patients receiving Isoniazid preventive therapy reported a rate of hepatitis lower than that previously reported. Of these, 11 patients (0.10% of those starting and 0.15% of those completing therapy) developed clinical hepatitis (Nolan et al., 1999).
Methyldopa: Methyldopa is an antihypertensive that is contraindicated in patients with active hepatic dysfunction. Periodic determination of hepatic function should be performed during the first 6-12 weeks of therapy. Occasionally, fever may occur within 3 weeks of methyldopa therapy, which may be associated with abnormalities in liver function test results or eosinophilia, necessitating discontinuation. In some patients, findings are consistent with those of cholestasis and hepatocellular injury. Rarely, fatal hepatic necrosis has been reported after use of methyldopa, which may represent a hypersensitivity reaction (Lee and Denton, 1989).
Oral contraceptives: Oral contraceptives can lead to intra hepatic cholestasis with pruritus and jaundice in a small number of patients. Patients with recurrent idiopathic jaundice of pregnancy, severe pruritus of pregnancy, or a family history of these disorders are more susceptible to hepatic injury. Oral contraceptives are contraindicated in patients with a history of recurrent jaundice of pregnancy. Benign neoplasm, rarely malignant neoplasm of the liver and hepatic vein occlusion has also been associated with oral contraceptive treatment (Edmondson et al., 1976).
Pyrazinamide: The well known adverse effect of this drug is hepatotoxicity. Hepatotoxicity is dose related and may occur any time during therapy. In the Centre for Diseases Control (CDC) update, 48 cases of hepatotoxicity were reported in association with a 2 month regimen of Rifampin-pyrazinamide for the treatment of latent tuberculosis. Thirty-seven patients recovered and 11 died of liver failure. Of the 48 reported cases, 33 (69%) occurred in the second month of therapy (CDC Update, 2003).
Rifampicin: Transient abnormalities in liver function are common in the initial stages of therapy. But in some cases it may cause severe hepatotoxicity, more so in those with pre-existing liver disease, forcing the physician to change treatment and opt for liver friendly treatment. Rifampicin causes transient elevations in hepatic enzymes usually within the first 8 weeks of therapy in 10 to 15% of patients, with less than 1% of the patients demonstrating overt rifampicin-induced hepatotoxicity. The occurrence of mortality associated with hepatotoxicity has been reported to be 16 in 500,000 patients receiving rifampicin. A higher incidence of hepatotoxicity has been reported in patients receiving Rifampicin with other anti tubercular agents and is estimated to be fewer than 4% (Kucers et al., 1987).
Statins/HMG-Co A reductase inhibitors (package inserts): The use of statins is associated with biochemical abnormalities of liver function. Moderate elevations of serum transaminase levels have been reported following initiation of therapy and are often transient. Elevations are not accompanied by any symptoms and do not require interruption of treatment. Persistent increases in serum trans- aminase levels occur in approximately 1% of patients and these patients should be monitored until liver function returns to normal after drug withdrawal. Active liver disease or unexplained transaminase elevations are contraindications to use of these drugs. Exercise caution in patients with a recent history of liver disease or in persons who drink alcohol regularly and in large quantities. Statins are among the most widely prescribed medications in the western world (Chalasani, 2005; Chalasani et al., 2004).
Valproic acid and divalproex sodium: Microvesicular steatosis is found with alcohol, aspirin, valproic acid, amiodarone, piroxicam, stavudine, didanosine, nevirapine and high doses of tetracycline. Prolonged treatment with methotrexate, isonizde, ticrynafen, perhexiline, enalapril and valproic acid may lead to cirrhosis. Valproic acid typically causes microsteatosis.
| Table 3: | Herbal products with known hepatotoxicity (Bateman et al., 1998; Gordon et al., 1995) |
 | |
This drug should not be given to patients with hepatic disease; exercise caution in patients with a prior history of hepatic disease. Those at particular risk include children younger than 2 years, those with congenital metabolic disorders or organic brain disease and those with seizure disorders treated with multiple anticonvulsants (Beyeler et al., 1997).
Herbs: The increasing use of alternative medicines also has led to many reports of toxicity. The spectrum of liver disease is wide with these medicines (Bateman et al., 1998; Gordon et al., 1995).
| • | Jaundice with high transaminase levels may occur after 2 months of use, but it disappears after stopping the drug |
| • | Chaparral is used for a variety of conditions, including weight loss, cancer and skin conditions. It may cause jaundice and fulminant hepatic failure |
| • | Chinese herbs (Jin bu huan [Lycopodium serratum], Inchin-ko-to [TJ-135], Ma-huang [Ephedra equisetina]) have been associated with hepatotoxicity (Table 3) |
Risk factors and Hepatotoxicity
Gender: It is well known that women are more vulnerable than men to the toxic effects of drugs in the liver, however gender differences have not always become apparent when large case series were analyzed (Andrade et al., 2005a). Regarding the clinic pathological expression of hepatotoxicity, the variety of chronic auto immune hepatitis that is induced by drugs is seen almost exclusively in women. Hepatotoxicity with certain medications such as nitrofurantoin, chlorpromazine, tetracycline, halothane and diclofenac has been reported more frequently in women (Andrade et al., 2005b). Female sex along with hepatocellular liver damage and increased total bilirubin levels on admission is suggested to be a risk factor for development of fulminant liver failure (Andrade et al., 2005a).
Age: Analysis of a cohort of patients with hepatic dysfunction, considered all drugs collectively suggested older age to be a risk factor to develop hepatotoxicity (Andrade et al., 2005a). A large Spanish cohort study reported the age-related pattern of liver damage resulting from Amoxicillin-Clavulanate (AC) treatment. According to this study older age is related to cholestatic/ mixed type of damage while younger age is associated with cytolitic damage (Lucena et al., 2006). Hepatocellular damage in the whole population was directly correlated with age and had the worst outcome (Andrade et al., 2005a).
Alcohol: Alcohol is capable of modulating the hepatotoxic potential of other drugs through CYP induction, inhibition, or substrate competition. Alcohol seems to have a dual effect on CYP2E1. During chronic regular intake, ethanol enhances acetaminophen hepatotoxicity by inducting CYP2E1, as well as susceptibility to liver damage from isoniazid, methotrexate, halothane and cocaine and perhaps to other drugs that are substrates for this microsomal isoform. During acute intake, however, substrate competition with acetaminophen occurs, actually decreasing the speed of metabolism of this drug to its toxic intermediate. However, this latter effect is partially counteracted by the ability of alcohol to slow the degradation of the CYP2E1 fraction, thus enhancing again the formation of the harmful metabolite once alcohol intake is interrupted. Alcohol also contributes to paracetamol hepatotoxicity by the direct inhibition of glutathione synthesis and through the malnutrition that frequently accompanies chronic alcoholism (Andrade et al., 2005b).
Smoking: Cigarette smoking was reported to be a risk factor for the development of hepatic dysfunction (Sun et al., 2006; Benowitz et al., 2003). Cigarette smoke contains thousands of structurally diverse chemicals that possess cytotoxic, genotoxic and tumorigenic activity. A toxic air pollutant formed by smoking such as acrolein was reported to induce hepatotoxicity through direct mitochondrial damage (Sun et al., 2006). Moreover, smoking may induce CYP isoform (CYP2E1) and could contribute to acetaminophen hepatotoxicity and alcohol-induced liver disease (Benowitz et al., 2003).
THERAPEUTIC APPROACHES
Early diagnosis of drug-induced liver reactions is essential to minimizing toxicity. Monitoring hepatic enzyme levels is appropriate and necessary with a number of agents, especially with those that lead to overt toxicity. For drugs that produce hepatotoxicity unpredictably, biochemical monitoring is less useful. ALT (Alanin Transferase) values are more specific than AST (Aspartate Transferase) values. ALT values that are within the reference range at baseline and rise 2- to 3-fold should lead to enhanced vigilance in terms of more frequent monitoring. ALT values 4-5 times higher than the reference range should lead to prompt discontinuation of the drug.
No specific treatment is indicated for drug-induced liver disease. Treatment is generally supportive and based on symptomatology. Other than different synthetic compounds several hundred plants have been examined for use in a wide variety of liver disorders. About 170 phytoconstituents isolated from 110 plants belonging to 55 families were stated to possess liver protective activity about 600 commercial herbal formulations with claimed hepatic protective activity are being marketed worldwide (Trease and Evans, 2002).
The first step of management of hepatotoxicity is to discontinue the suspected drug. Specific therapy against drug-induced hepatotoxicity is limited to the use of N-acetylcysteine in the early phases of paracetamol toxicity. L-carnitine is potentially valuable in cases of valproate toxicity. In general, corticosteroids have no definitive role in treatment. They may suppress the systemic features associated with hypersensitivity or allergic reactions. Management of protracted drug-induced cholestasis is similar to that for primary biliary cirrhosis. Cholestyramine may be used for alleviation of pruritus. Ursodeoxycholic acid may be used. Lastly, consulting a hepatologist is always helpful (Bass, 2003).
Management of paracetamol-induced hepatotoxicity
N-Acetylcysteine: The antidote for APAP hepatotoxicity is NAC (N-Acetylcysteine). It is recommended in all patients in whom the quantities of APAP ingested, serum drug levels, or rising aminotransferases indicate a risk of hepatotoxicity. Its use is also suggested in patients with acute liver failure when APAP ingestion is possible or even when knowledge of circumstances surrounding admission is inadequate (Polson and Lee, 2005).
When administered early (within 8 h of APAP ingestion), NAC limits the accumulation of NAPQI (N-acetyl-para-benzoquinoneimine/N-acetylbenzoquinoneimine) by directly binding to it, increasing glutathione stores and increasing sulfate conjugation (Lin and Levy, 1981). No deaths have been reported in larger studies in which NAC was administered within 10 h of APAP ingestion, regardless of serum levels (Smilkstein et al., 1991).
Oral charcoal: Oral activated charcoal is also useful if given within 4 h of APAP ingestion. It may be used beyond the initial 4 h in the presence of delayed gastric emptying or APAP absorption (e,g., with co-ingestants that reduce gut motility). It has been noted to adsorb APAP, resulting in reduced absorption of the drug. In patients with known or suspected APAP overdose who present within 4 h of ingestion, administration of activated charcoal is recommended as first-line therapy, even before NAC (Polson and Lee, 2005; Green et al., 2001). A study comparing the use of gastric lavage, syrup of ipecac and oral activated charcoal among 20 patients found that activated charcoal produced a greater lowering of mean serum APAP levels than other interventions (Underhill et al., 1990). It has the best risk-benefit ratio in comparison with other decontaminants (Brok et al., 2006). Oral activated charcoal is administered as a single oral dose of 1 g kg-1. There is no benefit to the use of divided doses.
Cimetidine: The use of cimetidine to treat APAP toxicity was based on the observation that it is also metabolized by the cytochrome P450 2E1 pathway, which would theoretically lead to competitive inhibition of the enzyme and reduce APAP metabolism to NAPQI. In an early study, however, Slattery et al. (1989) reported that the administration of 300 mg of cimetidine every 6 h to 13 subjects after 8 h of APAP ingestion did not alter APAP metabolism or APAP elimination or reduce alanine transferase or aspartate transferase levels. It was suggested that this lack of effect reflected the late administration of cimetidine after APAP ingestion (Burkhart et al., 1995).
Dialysis: The role of extracorporeal elimination in APAP intoxication is controversial and the data are scanty. Hemodialysis has been used in severe APAP hepatotoxicity as the drug is dialyzable. However, results have not shown that hemodialysis prevents or reduces the risk of hepatotoxicity (McBride and Rumack, 1992).
Prevention and treatment of acute hepatotoxicity caused by unpredictable (idiosyncratic) hepatotoxins: Because no specific antidotal treatments exist for the forms of toxicity that are caused by drug allergy or metabolic idiosyncrasy, prevention is paramount. Severe immunologically mediated or allergic hepatitis is generally considered an indication for steroid therapy, but only anecdotal reports support its use and there is scarce evidence of its benefits (Deleve and Kaplowitz, 2000). Management of acute non-immunologic hepatic injury consists of supportive and symptomatic treatment, the nature of which depends on the form of injury.
Treatment of idiosyncratic acute hepatocellular injury: Drug-induced hepatocellular jaundice has a potential case fatality rate of 10% or more. Accordingly, it warrants careful observation for evidence of impending hepatic failure. In the patient whose jaundice is not severe, whose prothrombin time is normal or negligibly prolonged and who has no clinical evidence of impending encephalopathy or coagulopathy, medical management can be simply supportive and the individual can be followed on an outpatient basis. Unless there is evidence of impending hepatic failure, a standard diet is appropriate, with no need to modify the protein or other components. Persistent anorexia may be managed by multiple small feedings and by providing fruits, vegetables and dairy foods rather than meat. Carbonated drinks, fruit juice and hard candy are usually well tolerated even when nausea is marked. There is no need to restrict physical activity, although patients should be advised to stay within limits of fatigability. The patient with measurable prolongation of prothrombin time and elevated bilirubin levels should be hospitalized (or observed very closely as an outpatient), particularly if there is persistent nausea and anorexia after the drug has been withdrawn (Gruchalla, 2000).
Treatment of acute cholestatic injury: Acute drug-induced cholestatic jaundice is rarely fatal. Over 99% of patients with cholestatic jaundice caused by erythromycin, chlorpromazine, amoxicillin-clavulanate, or anabolic steroids have survived the episode. There is no firm evidence that any therapeutic measures affect the rate of disappearance of drug-induced cholestasis. However, several anecdotal observations suggest that treatment with ursodeoxycholic acid increases the rate of return to normal status (Mork et al., 1997; O’Brien et al., 1996; Katsinelos et al., 2000) and in our view the effort is warranted. The most important aspects of treatment of cholestatic jaundice relate to the treatment of pruritus. Cholestyramine, which can offer relief, presumably traps elements involved in the itching. Other potentially useful agents include hydroxyzine, rifampin (Rifadin) and narcotic antagonists. There is no evidence that glucocorticoids provide symptomatic or other benefit in drug-induced cholestasis. Perhaps most important is an awareness that certain drug-induced cholestatic reactions can be mistaken for syndromes of anatomic biliary obstruction calling for surgical intervention, as has been seen with erythromycin and amoxicillin-clavulanate (Lewis and Zimmerman, 1999).
Management of chronic drug-induced hepatic disease: Treatment of the various syndromes of chronic hepatic disease that may be drug-induced mainly involves recognition of symptoms and withdrawal of the responsible agent. The lesion and syndrome of chronic hepatitis may be caused by a number of agents and by different mechanisms.
Chronic autoimmune hepatitis: Drug-induced chronic autoimmune hepatitis may resemble, to a striking degree, the form of chronic necroinflammatory disease dubbed “autoimmune” in origin. This type of injury has been reported following use of several agents, including nitrofurantoin, minocycline, methyldopa, diclofenac and pemoline, among others (Lewis and Zimmerman, 1998). Indeed, in any form of non viral chronic hepatitis, especially with autoimmune features, a drug should be suspected as the cause. Following withdrawal, improvement should become noticeable within 1 to 4 weeks. In some instances where injury fails to abate despite withdrawal of the drug, glucocorticoid therapy may be included.
Chronic cholestasis: Drug-induced chronic cholestasis is usually a sequel to acute cholestatic injury with loss of portal area bile ducts Vanishing Bile Duct Syndrome, (VBDS). Currently, there is no accepted therapy for the cholestatic process in patients with VBDS; however, ursodiol has been used successfully in a few reported patients who had received amoxicillin-clavulanate, chlorpromazine, prochlorperazine (improving pruritus and liver function tests) androgens, anabolic steroids and tetracycline (Mork et al., 1997; O’Brien et al., 1996; Katsinelos et al., 2000; Singh et al., 1996). Long-term treatment with ursodiol, 300 to 600 mg, has been required in some patients with VBDS to control the manifestations of cholestasis (O’Brien et al., 1996). This syndrome usually resolves spontaneously, although it may take several months to years and only a minority of these patients develop secondary biliary cirrhosis (Desmet et al., 1998).
Fatty liver, fibrosis and cirrhosis: Drug-induced macrovesicular fatty liver is a lesion that, per se, offers little threat. The steatosis may, as with methotrexate (MTX), be the forerunner of a more severe form of liver disease, namely cirrhosis. Serious hepatic disease, however, appears to occur only in patients who are alcoholics or obese diabetics. Aminotransferase testing is considered to be adequate for monitoring of patients with rheumatoid arthritis (Kremer et al., 1996; Lewis, 1997), juvenile rheumatoid arthritis (Hashkes et al., 1999) taking MTX.
Referral to liver transplantation centre/surgical care: No specific antidote is available for the vast majority of hepatotoxic agents. Emergency liver transplantation has increasing utility in the setting of drug-induced fulminant hepatotoxicity. Considering early liver transplantation is important. The Model for End-Stage Liver Disease score can be used to evaluate short-term survival in an adult with end-stage liver disease. This can help stratify candidates for liver transplantation. The parameters used are serum creatinine, total bilirubin, international normalized ratio and the cause of the cirrhosis (Bass, 2003).
FUTURE PERSPECTIVES
A large prospective database creation on drug-induced hepatotoxicity in collaboration with multidisciplinary and multicentric networks focused on the identification of bona fide cases following the same structural report form has been the very first step to provide insights into epidemiology and pathogenesis of drug-induced hepatotoxicity. This has allowed creating a pharmacoepidemiological culture in the attending physicians that become more alert in the detection of drug-induced hepatotoxicity and understanding of complex mechanism of drug-induced hepatotoxicity. Drug-induced hepatotoxicity in paediatric patients is an orphan field and there is obvious need to develop strategies to accomplish implementation of a specific network in paediatric patients (Hoofnagle, 2004; Pineiro-Carrero and Pineiro , 2004; Squires et al., 2006). It is hoped that a better understanding of hepatotoxicity mechanisms will provide the development of more specific and effective forms of hepatic therapy in the near future.
REFERENCES
- Andrade, R.J., M.I. Lucena, M.C. Fernandez, G. Pelaez and K. Pachkoria, 2005. Drug-induced liver injury: An analysis of 461 incidences submitted to the Spanish registry over a 10-year period. Gastroenterology, 129: 512-521.
CrossRefDirect Link - Bateman, J., R.D. Chapman and D. Simpson, 1998. Possible toxicity of herbal remedies. Scott-Med. J., 43: 7-15.
CrossRefDirect Link - Batt, A.M. and L. Ferrari, 1995. Manifestations of chemically induced liver damage. Clin. Chem., 41: 1882-1887.
Direct Link - Benowitz, N.L., M.J. Peng and P. Jacob, 2003. Effects of cigarette smoking and carbon monoxide on chlorzoxazone and caffeine metabolism. Clin. Pharmacol. Ther., 74: 468-474.
PubMedDirect Link - Beyeler, C., J. Reichen and S.R. Thomann, 1997. Quantitative liver function in patients with rheumatoid arthritis treated with low-dose methotrexate: A longitudinal study. Br. J. Rheumatol., 36: 338-344.
Direct Link - Brok, J., N. Buckley and C. Gluud, 2006. Interventions for paracetamol (acetaminophen) overdose. Cochrane Database Syst. Rev.
CrossRefDirect Link - Burkhart, K.K., N. Janco, K.W. Kulig and B.H. Rumack, 1995. Cimetidine as adjunctive treatment for acetaminophen overdose. Hum. Exp. Toxicol., 14: 299-304.
CrossRefDirect Link - Chalasani, N., H. Aljadhey, J. Kesterson, M.D. Murray and S.D. Hall, 2004. Patients with elevated liver enzymes are not at higher risk for statin hepatotoxicity. Gastroenterology, 126: 1287-1292.
PubMed - Chalasani, N., 2005. Statins and hepatotoxicity: Focus on patients with fatty liver. Hepatology, 41: 690-695.
PubMedDirect Link - Chang, C.C., M. Petrelli, J.F. Tomashefski and A.J. Mccullough, 1999. Severe intrahepatic cholestasis caused by amiodarone toxicity after withdrawal of the drug: A case report and review of the literature. Arch. Pathol. Lab. Med., 123: 251-256.
PubMed - Desmet, V.J., P. van Eyken and T. Roskams, 1998. Histopathology of vanishing bile duct diseases. Adv. Clin. Pathol., 2: 87-99.
PubMed - Edmondson, H.A., B. Henderson and B. Benton, 1976. Liver-cell adenomas associated with use of oral contraceptives. N. Engl. J. Med., 294: 470-472.
PubMedDirect Link - Egger, T., H. Dormann, G. Ahne, A. Pahl and U. Runge et al., 2005. Cytochrome p450 polymorphisms in geriatric patients: Impact on adverse drug reactions a pilot study. Drugs Aging, 22: 265-272.
PubMed - Gordon, D.W., G. Rosenthal, J. Hart, R. Sirota and A.L. Baker, 1995. Chaparral ingestion. The broadening spectrum of liver injury caused by herbal medications. JAMA, 273: 489-490.
PubMedDirect Link - Gruchalla, R.S., 2000. Clinical assessment of drug-induced disease. Lancet, 356: 1505-1511.
PubMedDirect Link - Gunawan, B. and N. Kaplowitz, 2004. Clinical perspectives on xenobiotic-induced hepatotoxicity. Drug Metab. Rev., 36: 301-312.
PubMedDirect Link - Hashkes, P.J., W.F. Balistreri, K.E. Bove, E.T. Ballard and M.H. Passo, 1999. The relationship of hepatotoxic risk factors and liver histology in methotrexate therapy for juvenile rheumatoid arthritis. J. Pediatr., 134: 47-52.
PubMedDirect Link - Hoofnagle, J.H., 2004. Drug-induced liver injury network (DILIN). Hepatology, 40: 773-773.
PubMedDirect Link - Tan, H.H., C.Y. Chang and P. Martin, 2009. Acetaminophen hepatotoxicity: Current management. Mount Sinai J. Med., 76: 75-83.
PubMedDirect Link - Ibanez, L., E. Perez, X. Vidal and J.R. Laporte, 2002. Prospective surveillance of acute serious liver disease unrelated to infectious, obstructive, or metabolic diseases: Epidemiological and clinical features and exposure to drugs. J. Hepatol., 37: 592-600.
PubMed - Ansari, J.A., M. Khan and M.A. Baig, 2009. Liver toxicity caused by synthetic drugs. J. Chem. Pharm. Sci., 2: 265-269.
Direct Link - Katsinelos, P., T. Vasiliadis and P. Xiarchos, 2000. Ursodeoxycholic acid (UDCA) for the treatment of amoxycillin-clavulanate potassium (Augmentin) -induced intra-hepatic cholestasis: Report of two cases. Eur. J. Gastroenterol. Hepatol., 12: 365-368.
PubMed - Kremer, J.M., D.E. Furst, M.E. Weinblatt and S.D. Blotner, 1996. Significant changes in serum AST across hepatic histological biopsy grades: Prospective analysis of 3 cohorts receiving methotrexate therapy for rheumatoid arthritis. J. Rheumatol., 23: 459-461.
PubMed - Larrey, D., 2002. Epidemiology and individual susceptibility to adverse drug reactions affecting the liver. Seminars Liver Dis., 22: 145-155.
PubMed - Lee, W.M. and W.T. Denton, 1989. Chronic hepatitis and indolent cirrhosis due to methyldopa: The bottom of the iceberg. J. South Carolina Med. Assoc., 85: 75-79.
PubMed - Lewis, J.H., 1997. Monitoring for methotrexate hepatotoxicity in patients with rheumatoid arthritis: Another hepatologists perspective. J. Rheumatol., 24: 1459-1460.
PubMed - Lewis, J.H. and H.J. Zimmerman, 1999. Drug and chemical induced cholestasis. Clin. Liver Dis, 3: 433-464.
PubMed - Lin, J.H. and G. Levy, 1981. Sulfate depletion after acetaminophen administration and replenishment by infusion of sodium sulfate or N-acetylcysteine in rats. Biochem. Pharmacol., 30: 2723-2725.
PubMed - Lucena, M.I. R.J. Andrade, M.C. Fernandez, K. Pachkoria and G. Pelaez et al., 2006. Determinants of the clinical expression of amoxicillin-clavulanate hepatotoxicity: A prospective series from Spain. Hepatology, 44: 850-856.
PubMedDirect Link - Morelli, S., V. Guido and P. De Marzio, 1991. Early hepatitis during intravenous amiodarone administration. Cardiology, 78: 291-294.
PubMed - Mork, H., O. Al-Taie, O. Klinge and M. Scheurlen, 1997. Successful therapy of persistent androgen-induced cholestasis with ursodeoxycholic acid. Z. Gastroenterol., 35: 1087-1091.
PubMed - Nolan, C.M. S.V. Goldberg and S.E. Buskin, 1999. Hepatotoxicity associated with isoniazid preventive therapy: A 7-year survey from a public health tuberculosis clinic. JAMA, 281: 1014-1018.
PubMed - O'Brien, C.B., D.S. Shields, S.H. Saul and R. Reddy, 1996. Drug induced vanishing bile duct syndrome: Response to ursodiol. Am. J. Gastroenterol., 91: 1456-1457.
PubMed - Park, B.K., M. Pirmohamed and N.R. Kitteringham, 1998. Role of drug disposition in drug hypersensitivity: A chemical, molecular and clinical perspective. Chem. Res. Toxicol., 11: 969-988.
PubMed - Polson, J. and W.M. Lee, 2005. AASLD position paper: The management of acute liver failure. Hepatology, 41: 1179-1197.
CrossRefPubMedDirect Link - Ramachandran, R. and S. Kakar, 2009. Histological patterns in drug-induced liver disease. J. Clin. Pathol., 62: 481-492.
CrossRefDirect Link - Nelson, S.D., 1990. Molecular mechanisms of the hepatotoxicity caused by acetaminophen. Semin. Liver Dis., 10: 267-278.
CrossRefPubMedDirect Link - Singh, C., P. Bishop and R. Wilson, 1996. Extreme hyperbilirubinemia associated with the use of anabolic steroids, health/nutritional supplements and ethanol: Response to ursodeoxycholic acid treatment. Am. J. Gastroenterol., 91: 783-785.
PubMed - Slattery, J.T., T.I. McRorie, R. Reynolds, T.F. Kalhorn, E.D. Kharasch and A.C. Eddy, 1989. Lack of effect of cimetidine on acetaminophen disposition in humans. Clin. Pharmacol. Ther., 46: 591-597.
CrossRefPubMedDirect Link - Smilkstein, M.J., A.C. Bronstein and C. Linden, 1991. Acetaminophen overdose: A 48-hour intravenous nacetylcysteine treatment protocol. Ann. Emerg. Med., 20: 1058-1063.
PubMed - Squires, Jr. R.H., B.L. Shneider and J. Bucuvalas, 2006. Acute liver failure in children: The first 348 patients in the pediatric acute liver failure study group. J. Pediatr., 148: 652-658.
PubMed - Sun, L., C. Luo, J. Long, D. Wei and J. Liu, 2006. Acrolein is a mitochondrial toxin: Effects on respiratory function and enzyme activities in isolated rat liver mitochondria. Mitochondrion, 6: 136-142.
PubMed - Tahaoglu, K., G. Atac and A.T. Sevim, 2001. The management of anti-tuberculosis drug-induced hepatotoxicity. Int. J. Tuberculosis Lung Dis., 5: 65-69.
PubMed - Underhill, T.J., M.K. Greene and A. Dove, 1990. A comparison of the efficacy of gastric lavage, ipecacuanha and activated charcoal in the emergency management of paracetamol overdose. Arch. Emerg. Med., 7: 148-154.
PubMed - Walgren, J.L., M.D. Mitchell and D.C. Thompson, 2005. Role of metabolism in drug-induced idiosyncratic hepatotoxicity. Crit. Rev. Toxicol., 35: 325-361.
PubMed - Zimmerman, H.J., 1981. Effects of aspirin and acetaminophen on the liver. Arch. Internal Med., 141: 333-342.
Direct Link