
Abdul Arif Khan
Microbiology Unit, Department of Pharmaceutics, College of Pharmacy, King Saud University, P.O. Box 2457, Riyadh, 11451, Saudi Arabia
Journal of Biological Sciences
Year: 2010 | Volume: 10 | Issue: 4 | Page No.: 291-305







ABSTRACT
Everything that has originated in this world will have to ends its journey of life. However in biological system this journey is previously programmed and this programming maintains the cells to carry out its all vital activities and after certain time, this journey ends to its final destination, i.e., death. However in case of simple unicellular organism, this journey is too simple and is controlled by the programming in the organism it self. But in case of large multi-cellular organism, where the cells are the building block of multi-cellular architecture, each and every cell is in continuous contact with other cells. Journey of the cell is influenced by its neighbors, so this programming becomes too much complicated in case of higher organisms. Usually when cell die, the death may take place by three means. First, the death of cell after completing its journey of life by Apoptosis. Second, death arises due to injury i.e., necrosis. This is simply like, death of a person, after aging and before aging by accident. Third situation is intermediate to both that is paratopsis. Within all these types of cell death, only apoptosis is truly programmed cell death that takes place due to execution of cell death program. This programming is regulated by the various cellular messenger, stimulator and positive and negative regulator of apoptosis. This review covers basic regulation of apoptosis machinery. Understanding of these pathways can aid in the understanding of molecular biology of cancer, AIDS and various other diseases.
PDF Abstract XML References Citation
How to cite this article
Abdul Arif Khan, 2010. Intracellular Mechanisms of Apoptosis. Journal of Biological Sciences, 10: 291-305.
DOI: 10.3923/jbs.2010.291.305
URL: https://scialert.net/abstract/?doi=jbs.2010.291.305
DOI: 10.3923/jbs.2010.291.305
URL: https://scialert.net/abstract/?doi=jbs.2010.291.305
INTRODUCTION
A great Chinese philosopher Confucius said we don’t know life; how can we know death. This is a philosopher’s view on life and death. However, life in an organism is a result of complex biochemical reactions that leads to performance of all the vital activities in a cell. These all biochemical reactions work in a proper manner to maintain survival of a cell. However these biochemical reactions don’t works indefinitely in a cell. Performance of these vital activities depend on previous programming in the organism itself and after the end of this program, cell looses its vital activities and become, dead. In case of multi cellular organism this programmed death is normal consequence because if a cell will not die after certain time period with continuous formation of new cells, leading to the expansion in the number of cells resulting in disturbance of cellular homeostasis. Similarly, in case of programmed cell death not happening, the embryo will not take its proper shape and will develop into only a mass of cell. Beside it, when a pathogen attacks an individual, it tries to control its host and replicate inside or on it. In case of multi-cellular organisms, pathogen grows in some cells of host and host try to remove these infected cells to prevent further infection. This removal of infected cells is also due to programmed cell death and is just like surgery of diseased part at cellular level. So this special programmed cell death take place for development, homeostasis, defense, aging etc.
When a cell dies, death may take place due to three means. First, by the regular programming in a cell and in such case, cell die with out releasing its intracellular constituents in environment. Second, by injury in which the intracellular constituent gets released in surrounding environment and inform the surrounding environment about injury. Both of the above mentioned type of cell death shows some of its characteristic morphological feature. Such as in programmed cell death, nuclear fragmentation, chromatin condensation occurs. Cell shrinks and disintegrates to form small bodies, which are engulfed by adjacent cells without releasing its intracellular constituents. Whereas in death of injured cell, nuclear fragmentation and chromatin condensation don’t takes place and cell swell and burst to release its intracellular constituents in surrounding environment, while a third type of cell death shows morphological pattern, intermediate to both above described type of cell death. This review tries to focus on the pathways of responsible for the programmed cell death.
Human beings are infected by variety of organisms from variety of sources (Jain et al., 2008; Bhatnagar et al., 2007a, b). During infection, pathogens adapt themselves, according to the environment including host where they normally reside and subsist. In doing so, they change themselves; according to environment, as well as try to change their environment i.e., host, according to them for effective exploitation of host. So, during establishment of infection, a continuous interplay of adaptation between host and pathogen takes place. And when the infection is by a virus, this interplay becomes a striking feature for development of infection. Because virus are simplest entities, they have nothing in itself, but the environment surrounding gives them chance to grow and to becomes a dangerous pathogen. So the pathogenic capability of a virus depends on effective exploitation of host cell and this effective exploitation includes control on the programmed cell death of host.
Programmed cell death: Cell death may by due to aging, injury or other reasons. But in each and every case, morphological pattern of cell death differ. Cell death may occur due to previous programming and this programmed cell death called as apoptosis, which shows nuclear fragmentation chromatin condensation, apoptotic bodies formation, these apoptotic bodies are engulfed by adjacent cell with out eliciting any inflammatory response. The next type of cell death due to injury, in which nuclear fragmentation, chromatin condensation and apoptotic bodies formation do not occur, but cytoplasmic vacoulation and mitochondrial swelling are characteristic features. In this type of cell death, the cell swells and releases its intracellular constituents to induce inflammatory response. This is called necrosis. A third type of cell death, in which nuclear fragmentation and apoptotic bodies formation are absent, but cytoplasmic vacoulation is a prominent feature and chromatin condensation may or may not occur. This is called as paratopsis. This process of paratopsis occur in some autophagic, cytoplasmic and neurodegenerative cell death (Sprandio et al., 2000). Besides, apoptosis may also take place from a combination of deficient cell cycle check points and cellular damage, this type of apoptosis is known as mitotic catastrophe (Castedo et al., 2004). Moreover, total cell cycle arrest also resulted in aging process and is known as cellular senescence (Vicencio et al., 2008).
In all these above described cell deaths, apoptosis is totally programmed cell death. Apoptosis, firstly discovered by Carl Vogt in 1842, but their discovery remained dormant for more than a hundred year until Kerr, Willie and Currie rediscovered this programmed cell death in 1972 and coined the term apoptosis inspired from the word apoptotic bodies, mean to fall away from or the falling of leaves in the autumn from deciduas trees. Apoptosis is evolutionary conserved from of cell death, which is conserved throughout evolution from nematode to man. The key components of apoptosis machinery seem to be conserved between human and nematode (Peter et al., 1997; Reed, 2000).
In the Nematode Caenorhabditis elegans, 3 genes ced-3, ced-4 and ced-9 directly regulate apoptosis. Both ced-3 and ced-4 are killer genes and induce apoptosis while ced-9 is survival genes and protect cell from apoptosis. ced-3 is homologous to caspase-3 in human and ced-9 is homologue of human Bcl-2, whereas ced-4 work as a bridge between ced-3 and ced-9 to induce apoptosis and is homologous to human death enzyme Apaf-1 (Peter et al., 1997; Sheshagiri and Miller, 1997). For a convenient study apoptosis can be divided into four phases.
Stimulus: This stimulus is a signal to end journey of life. In normal consequences, this stimulus arises due to autonomous programming in cell or this programming may be induced either by external signals which are transferred through, surface receptor present on a cell, for example, CD95/CD95 ligand or TNF/TNF receptor or by internal signals, originated through action of certain drug, toxin, radiation etc.
Signal transaction: Once these signals of apoptosis arise, these are delivered from external and internal source to its detection machinery, which are trans-membrane proteins or metabolic state. This detection machinery detects the signal; transduce it to the cell death effecter machinery for their activation. During signal transduction, variety of protein interacts with each other to transduce the signal. Basically three types of protein act during cell death signal transduction (1) Signal acceptor, (2) Adapter (Intermediate protein) and (3) Death effecter machinery activator. Signal acceptor, accept, signal, Death effecter machinery activator, activate, death machinery whereas adapters work as a bridge between both of two. These all protein interacts with each other due to presence of certain homologous domains in them. Besides, many other proteins work as silencer or inducers of this signal transduction pathway.
Effecter machinery action: After transfer of death signal to, death effecter machinery, it resulted in its activation. Death effecter machinery comprises various cellular proteases (Caspase) that acts on its substrates and degrade them, during overall action of effecter machinery various positive and negative regulator of effecter machinery act to modulate its action. Thus, effecter machinery action causes demise of a cell that ultimately leads to apoptosis.
Post mortem phase: During this phase various cellular endonucleases are activated, that degrades the cellular DNA, resulted in chromatin condensation as well as degradation of cytoskeleton that results in shrinkage of cell and ultimately lead to formation of apoptotic bodies. Apoptotic bodies are recognized and engulfed by adjacent cells by phagocytosis (Vaux and Strasser, 1996).
So, during all these phases of apoptosis, typical morphology of apoptosis is caused by specific protease. These proteases are executioner of entire apoptotic machinery. Apoptotic machinery comprises specific class of Cystiene Aspartyl specific protease called as caspase. Although researchers firstly identified first caspase as Interleukin 1 β converting enzyme (ICE or caspase 1) but this caspase has major role in inflammation and the first caspase found to be involved in apoptosis was C. elegans ced-3 gene product. Its human homologue is known as caspase-3, caspase show similarity in their amino acid sequences and structure. All caspase are present normally as zymogen (proenzyme) that consists of three parts. NH2 terminal domain, a 20 kD large subunit and 10 kD small subunit. Activation of these procaspase requires cleavage at an internal conserved Asp residue to generate a heterodimer of large and small subunit and ultimate association of two large and two small subunits to generate a hetero tetramer (active enzyme). These active caspase or enzyme generates various different substrate specificities by association of different large and small chains. These active enzymes may inactivate proteins required for cell survival, such as active caspase may degrade ICAD (Inhibitor of caspase activated deoxyribonuclease) and lead to activation of deoxyribonuclease and degrades DNA or may directly degrade proteins required for cell structure maintenance, DNA repair, mRNA splicing and DNA replication (Thornberry and Lazebnik, 1998).
For sake of convenience caspase are grouped into two groups, Initiator and effector caspase. Initiator caspase initiate, apoptosis machinery activation, while effecter caspase are the part of apoptosis machinery that activates due to Initiator caspase (Nunez et al., 1998).. Initiator caspase consist of large NH2 terminal pro-domain to interact with large group of stimulator as well as positive and negative regulator of cell death, while effecter caspase have small NH2 terminal pro-domain, that shows its restricted activation interactions (Barisic et al., 2003).
CASPASE ACTIVATION
Usually various models are available to focus on caspase activation mechanism but some most popular models include induced proximity model, hypothesized that procaspase have low enzymatic activity and this enzymatic activity is enough for auto processing of procaspase and generation of active caspase. An Apoptotic stimulus induces over expression of procaspase that lead to their artificial cross linkage and activation (Thornberry and Lazebnik, 1998; Muzio et al., 1997).
The second most popular model is facilitated auto catalysis model, that postulates, all component of caspase activation are present in a cell, but only apoptotic stimulation facilitate their activation by conformational change and ultimate auto catalysis of procaspase to generate active caspase. This model is supported by the facts that various regulators of apoptosis remain inactive, but apoptotic stimulation facilitates their activation and ultimate activation of caspase by auto catalysis (discussed in domain of apoptosis).
Third model of caspase activation is compartmentalization model, it assumes that normally all the components required for caspase activation remains inside a cell, but in different part or compartments. During the apoptotic response these all sequestered proteins interact with each other and cause cell death. However all these models are right for apoptotic response with certain limitation and not a single model is applicable for all the apoptotic response.
Till now, at least 14 caspase have been identified in mammals in which 11 are in humans. Within all these known caspase, caspase 2, 8, 9 and 10 work as initiator caspase and caspase 3, 6 and 7 work as effecter caspase while caspase 1, 4 and 5 have no direct involvement in apoptosis, they are IL-1β converting enzyme and involved in inflammation. Although, we could not explore our knowledge about initiator caspase activation and only mechanism of effecter caspase activation (Caspase-7) has been explained. This caspase was shown to consist of large and small subunit. Large and small subunit monomers form a homodimer by hydrophobic interaction. The protein chains of each monomer form 4 active loops (L1-L4 and L1'-L4'), during activation of caspase, rearrangement in these loops occurs to assume active confirmation by intra chain cleavage in L2 loop at catalytic cystiene and L2' loop. This cleavage leads to the formation of substrate binding groove (by L2' and L2) in active caspase. Because the structure of L2', L2 and L4 loop are generally conserved, it is assumed, that same mechanism activate other caspase (Riedl and Shi, 2004).
However, we could not explore our knowledge on initiator caspase activation at structural level, but the pathways through which, stimulation lead to execution of apoptosis in cell, have been explored very much. Apoptotic stimulation may arise either from inside or from outside the cell. These two different types of stimulation lead to two different pathways of apoptosis. However, this is not always the case, in some instances, cross talk between both internal and external apoptotic pathways takes place. These internal and external pathways are usually called as intrinsic or extrinsic pathways, respectively (Table 1).
| Table 1: | A brief analysis of apoptosis pathways, with its signal transduction and regulation, however this table includes only some member of apoptosis pathways |
 | |
| *Crosstalk between intrinsic and extrinsic pathways may occur. CED: Cell death abnormal | |
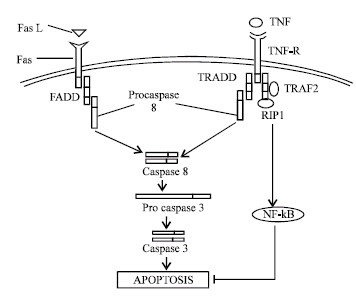 | |
| Fig. 1: | Extrinsic pathways of apoptosis with major pathways, however this figure is over simplification of entire extrinsic pathways of apoptosis. In all figure } denotes to inhibition of reaction and → denotes to comppletion of reaction |
Extrinsic pathways: This is also called as receptor mediated apoptosis pathway, because execution of apoptosis by this pathway relies upon interaction of specific death receptor with its ligand. These receptors are located on surface of the cells and induce apoptosis by their intracellular domains (Fig. 1). Basically these receptor are of TNF receptor family and consist of cysteine rich motifs repeated two to six times, most important of these are Fas (APO1/ CD95) and TNFR. Both these proteins carry a unique cytoplasmic death domain (DD), through which they interact with other proteins containing DD by homotypic interaction. These proteins are called as adaptor proteins, which transmit signal of apoptosis inside the cell.
In case of Fas mediated apoptotic pathway, Fas interaction with Fas ligand lead to conformationals alteration in Fas and ultimate interaction of Fas with FADD (Fas associated death domain) or MORT1 (Mediator of receptor induced toxicity) by the homotypic DD interaction and this interaction of death receptor and adaptor proteins lead to activation of initiator caspase 8. Thus, complex formed by interaction of above mentioned proteins lead to formation of DISC (Death inducing signaling complex). It activates effector caspase-3 and ultimate apoptosis (Muzio et al., 1997; Zhang et al., 1998). While in case of apoptosis mediated by TNFR1, through it's interaction with TNF also depends on conformational alteration in TNFR1. However adaptor protein differ in case of TNFR signaling, here another adaptor protein TRADD (TNF receptor associated DD) reacts with FADD to transfer extra cellular signal to apoptotic machinery. This TRADD act both as a positive as well as negative regulator of apoptosis, because interaction of TRADD with TNFR1 lead to activation of both initiator caspase and apoptotic machinery while in the absence of TNFR1 mediated apoptotic signal, TRADD activate NF-kB (Transcription factor) that initiate production of anti apoptotic proteins (Hsu et al., 1995; Gupta, 2003).
In addition to Fas and TNFR1, DR3, DR6 (Decay receptor) are also death domain containing proteins and interact with adaptor protein TRADD to induce apoptosis (Chinnaiyan et al., 1996; Pan et al., 1998). Ligand for Decay receptors are called as TRAIL (TNF related apoptosis inducing ligand). However, engagement of TNF receptor with their appropriate ligand lead to conformational alteration in the receptor molecule, involving trimerization (By three receptor monomer) of the receptor molecule because most of the ligand molecules are trimers. But some researchers believe that, the receptor are actually dimeric, they give evidence that unliganded receptor are dimeric, such as CD27 (TNFR family member) is a dimer formed by disulfide bond between two chains. Some ligands of TNFR family (NGFR, nerve growth factor receptor) are also dimeric. Beside it, single antibody to TNF receptor can not block signal transduction through trimeric TNF receptor, while if the receptor are actually trimeric, then it should block signal transduction by inhibiting one monomer (Beutler and Bazzani, 1998). Perhaps, these receptors are morphologically trimeric but functionally dimeric molecule. TNFR1 is a novel molecule that can signal for both, apoptosis and proliferation. This different signaling depends on interaction of adaptor proteins TRADD to cell proliferating or apoptosis inducing molecules. Such as interaction of TRADD to RIP1 (Receptor inducing protein 1) and TRAF2 lead to formation of complex I, that inhibit activation of caspase-8 and apoptosis, on the other hand complex I induces formation of NF-kB and ultimately induce proliferation of the cell, while, interaction of TRADD to FADD lead to formation of complex II and ultimate activation of caspase-8 and apoptosis (Micheau and Tschopp, 2003).
In various cases, this TNF receptor regulates apoptosis induced by other member of this family. Such as, TNFR2 induce increase expression of pro-apoptotic molecule Bax and inhibit anti apoptotic molecule FLIP [FLICE (Fas associated DD containing IL-1β converting enzyme) inhibitory protein] and induce apoptosis by Fas (Elzey et al., 2001). TNF α induces expression of Fas on cell surface in presence of NF-kB (Rel A) and stimulate Fas dependent apoptosis by T cells (Zheng et al., 2001a, b). However T cytotoxic cells (CTL) also use protease Granzyme B for induction of apoptosis in target cells. These cells create pores in target cells (By Perforins) and GrB enter inside target cell, through these pores and cleave 45 kDa subunit of DNA fragmentation factor (DFF 45). This cleavage resulted in DNA fragmentation and induction of apoptosis in target cells (Sharif-Askari et al., 2001). TNF α may depend on various other pathways for inducing apoptosis by TNF receptor. In liver cells TNF-α induce prodeath molecule Box/Bak and ROS generation and activate JNK kinase protease cathapsin B, Sphingomylinase. These all events induce apoptosis in liver cells (Ding and Yin, 2004). During induction of apoptosis by receptor mediated pathway, various proteins act to regulate other members of the pathway and these proteins will be discussed during domains interaction.
Intrinsic pathway: This pathway is so called because, signal for this pathway arise from inside the cell but in some instances external pathway may also crosstalk with intrinsic pathway for its activation. Signal for this pathway arises from diverse stimuli, such as DNA damage, growth factor withdrawal, cytotoxic drug and cell cycle irregularities etc. These all signals lead to activation of intrinsic pathways with the help of mitochondria. How mitochondria can kill a cell? To explain this question, three general mechanisms have been proposed.
First: Mitochondria inhibit normal respiratory activity during apoptotic signal. Such as, it disrupts electron transfer, oxidative phosphorylation and ATP production and ultimately leads to apoptosis. But loss of ATP production is a late event in apoptosis, so this mechanism can not work as apoptosis inducing pathway.
Second: Mitochondria carry various apoptosis regulatory proteins in itself. During the intrinsic apoptosis pathway, it releases these proteins that activate caspase and apoptosis occur.
Third: Mitochondria produce various super oxide anions during transfer of electron to molecular oxygen. Apoptotic signal increase production of these superoxide anion manifolds, leading to demise of a cell (Green and Reed, 1998).
Mitochondria release various proteins in response to intrinsic apoptotic stimuli (Fig. 2). In which, most important are cytochrome C, AIF (Apoptosis inducing factor), Endonuclease G and various other apoptosis regulatory proteins of Bcl-2 family. Once cytochrome C releases from mitochondria, it binds to Apaf-1 (Apoptotic protease activating factor-1). Apaf-1 is a cytosolic protein, containing caspase activating recruitment domain (CARD), WD-repeat and a nucleotide binding domain. Apaf-1 normally binds to dATP/ATP through its nucleotide binding domain weakly, but after binding of cytochrome C to WD-repeat, this interaction increases manifold. Now multiple cytochrome C/Apaf-1/dATP/ATP complex oligomerises to form apoptosome. CARD domain of Apaf-1 exposed during this oligomerization and binds to procaspase-9, through its CARD domain. Interaction of apoptosome with procaspase-9 induces its cleavage and activation of caspase-9 (Initiator caspase) (Saleh et al., 1999). Activation of caspase-9 causes activation of effecter caspase-3 and ultimate apoptosis (Li et al., 1997). So, caspase-9 work as initiator caspase of intrinsic pathway but some proteins, like Nod 1 carries CARD domain in itself and directly activate caspase-9 resulted in caspase-9 mediated apoptosis (Inohara et al., 1999).
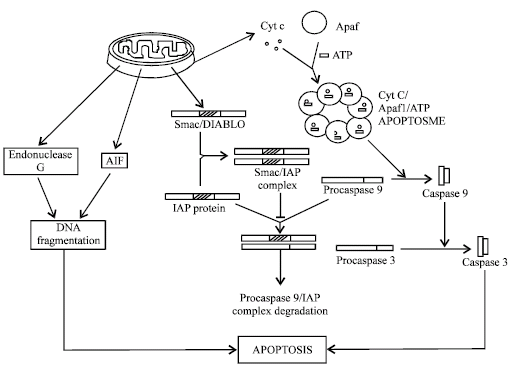 | |
| Fig. 2: | Inttrisic pathways of apoptosis, these depend of secretion of various apoptotic regulatory proteins from mitochondria |
Other apoptosis inducing proteins released from mitochondria are AIF and Endonuclease G. AIF is a 57 kDa flevoprotein resembling bacterial oxidoreductase, while endonuclease G is a 30 kDa nuclease in mitochondria. AIF function to maintain, mitochondrial physiology and endonuclease G act to remove RNA primers during mitochondrial DNA replication. In response to apoptotic stimulation, both proteins released from mitochondria to nucleus and cause DNA fragmentation, chromatin condensation that ultimately leads to apoptosis in a caspase independent manner. Apart from it, mitochondria also releases various apoptosis regulatory proteins, such as Smac (Second Mitochondria derived activator of caspase)/DIABLO (Direct inhibitor of apoptosis (IAP)-binding protein with low pI). Smac/ DIABLO protein bind to IAP family (Inhibitor of apoptosis) through its four amino acids Ala-Val-Arg-Ile sequence. This AVRI sequence binds to BIR (Baculovirus inhibitor of apoptosis repeat) domain of IAP. Normally, these IAP proteins bind to procaspase-9 and inhibit its activation and in some instance its degradation in proteosome. But after binding of IAP to Smac/ DIABLO, procaspase became free to be activated by apoptosome, resulted in apoptosis. Various other Bcl-2 family member also act to regulate apoptosis mediated by mitochondria, these are discussed separately in Bcl-2 domain (Wang, 2001).
Some apoptosis regulator, regulate both intrinsic and extrinsic pathway, such as protein kinase C induce caspase-9 activation, while inhibiting activation of caspase 2 and 8, so it act as a positive regulator of intrinsic pathway, while a negative regulator of extrinsic pathway (Basu and Miura, 2002). So, various proteins interact with each other to regulate entire apoptotic machinery. These interactions depend on presence of homologous amino acid sequence in each regulator. Moreover, interaction within these proteins mediated by domain (Amino acid sequence) presented within them, including death domain (DD), Death effecter domain (DED), Caspase activated recruitment domain (CARD), Bcl-2 homology (BH) domain of Bcl-2 family, BIR domain of IAP and in some instances NB-ARC domain (Nucleotide binding oligomerization domain) of Apaf-1, through which Apaf-1 bind to dATP/ATP. Protein with similar domain may interact with each other and transduce apoptotic signals to effecter machinery. Further article discusses each domain separately with their involvement in apoptosis regulation.
Death Domain (DD): Death domains are so called because, it transduce death signals. These domains belong to death domain superfamily, comprising DD, DED and CARD. These all three families of death domain superfamily show striking structural similarities and common evolutionary lineage. DD and CARD are most dissimilar to each other, while DED lies in the middle of the above two. Death domain of one protein interacts with death domain of others by homotypic interactions and
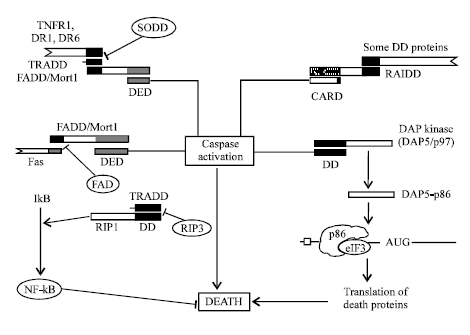 | |
| Fig. 3: | Role of different death domain containing proteins in regulation of apoptosis |
transduces effective signals (Fig. 3). Structurally DD consist of six α helix (Weber and Vincenz, 2001). Various apoptosis stimulator, adaptor, regulator carries DD in them, such as TNFR family receptor carries DD in them. Previously discussed Fas, TNFR1, DR3, DR5, DR6, all carries DD in their cytosolic region, upon activation with appropriate ligand their DD interact with special adaptor proteins containing DD. Such as, Fas bind to FADD through homotypic DD interaction. Adaptor protein FADD carries DD in conjunction with DED, so after binding of FADD to Fas by homotypic DD interaction, DED of FADD can interact with various DED containing caspase. These interactions resulted in activation of caspase and apoptosis.
However some proteins, such as FAP-1 (Fas associated phosphatase) can bind to Fas and inhibit its interaction to FADD, resulted in inhibition of Fas mediated apoptosis (Yanagisawa et al., 1997). Similarly, TNFR1, DR3, DR6 interact with another adaptor protein TRADD through its homotypic DD interaction. TRADD carries a single DD, through which it interact with above mentioned receptors by homotypic DD interaction, simultaneously it interact with DD of FADD or RIP-1, resulted in activation of caspase by FADD or activation of NF-kB by RIP-1 (Hsu et al., 1995; Ting et al., 1996). An apoptosis regulatory protein, SODD (Silencer of death domain) is a 60 kDa protein that can bind to DD of TNFR1 and DR3 and inhibit its interaction with TRADD, resulted in inhibition of receptor mediated apoptosis (Jiang et al., 1996).
Similarly another receptor inhibiting protein (RIP-3) can bind to RIP-1 and inhibit RIP-1 mediated NF-kB activation (Sun et al., 2002). Apart from it, various other DD containing adaptor proteins transmit apoptotic stimulation from receptor to the effecter machinery, such as RAIDD, which consist of DD in combination with CARD, can activate other CARD containing caspase through homotypic domain interaction (Duan and Dixit, 1997). Similarly another adaptor protein CRADD carries dual domain, one is caspase homology domain to interact with caspase-2, another is DD, which interact with RIP-1 or other DD containing protein (Ahmed et al., 1997). So, these all adaptor proteins interact with various other proteins through their different domain or by same domain through multivalent domain interaction. Some kinase, such as DAP (Death associated protein) kinase, can also induce apoptosis by homotypic DD interaction. DAP kinase remain as p97 protomer containing DD. When this protomer interact with other DD containing caspase, this protomer cleaved to generate p86 subunit, this p86 subunit act as a cofactor for eukaryotic initiation factor (eIF3) and it enhances translation of various death proteins, Apaf-1, c-myc etc. and resulted in apoptosis (Kimchi, 2002).
Death Effecter Domain (DED): DED also comprises conserved 6 α helix of DD super family. Within these 6 α helix, some conserved hydrophobic residues are normally present. Such as, negatively charged residue (Glutamic acid, Aspartic acid, Asparagine) at position 19 and Arg-X-Asp-Leu (R-X-D-L) motif is found at position 78-81 in nearly all DED proteins. Moreover these DED play major roles in regulating apoptosis. For example, previously described FADD carries both DD and DED and regulate apoptosis mediated by TNFR and Fas (Tibbetts et al., 2003). Beside it, various other proteins have DED, including caspase 8 and 10 (Interact with FADD through DED) and some inhibitor of apoptosis proteins such as FLIP, PEA15 etc. FLIP [FLICE (Caspase-8) inhibitory protein] can bind to FADD or caspase-8 through its DED and inhibit apoptosis mediated by caspase-8 and Fas. Some viruses also encode homologous proteins called vFLIP while cellular homologues are called as cFLIP. FLIP also induces NF-kB activation thus inducing transcription of anti apoptotic proteins resulted in inhibition of apoptosis (Tibbetts et al., 2003; Tschopp et al., 1998). Another DED containing protein is PEA-15 (Phospho protein enriched in astrocytes). PEA-15 inhibits apoptosis mediated by Fas or TNF-α, in a manner similar to FLIP. PEA-15 has also been shown to be implicated in oncogene Ras activation. So PEA makes two hits to initiate cancer. First hit by inhibiting apoptosis and second by promoting cell cycle (Tibbetts et al., 2003; Ramos et al., 2000). DEDAF is a novel protein, it does not have DED, but it interact with several proteins carrying DED. DEDAF (DED associated factor) interact with FADD, procaspase-8, procaspase-10 in cytosol and another DED containing protein DEDD in nucleus. DEDD enhances formation of DISC and death receptor mediated apoptosis. But in nucleus it re localize to DEDD to control nuclear event of apoptosis (Zheng et al., 2001a, b).
This DEDD (DED containing DNA binding protein) is normally found in cytoplasm but during receptor mediated apoptosis, it translocate to nucleus. Here it localize with UBF, a basal transcription factor for RNA Polymerase I. Due to all these events, it inhibit transcription and shut down biosynthetic activity that lead to apoptosis (Stegh et al., 1998). It has also been shown that DEDD bind to caspase-3, intermediate filament and activate caspase-6 and resulted in apoptosis (Tibbetts et al., 2003) (Fig. 4).
How DED can cause apoptosis, to solve this enigma some researchers demonstrated that DED containing proteins oligomerize to form filament like structure called as DEF (Death effecter filament) which in turn activate procaspase zymogen, resulted in apoptosis (Siegel et al., 1998).
Caspase Activated Recruitment Domain (CARD): CARD domain also belong to death domain superfamily and structurally consist of six α-helices, it interact with other CARD containing caspase. CARD proteins play major roles in regulation of apoptosis, inflammation and NF-κB signaling For the sake of simplicity, CARD family proteins have been divided in four subfamilies.
NBD-CARD: These proteins consist of a nucleotide binding domain and a CARD domain. Beside it, NBD-CARD also consists of certain repeat, such as leucine rich repeat or WD-repeat. This group consists of various
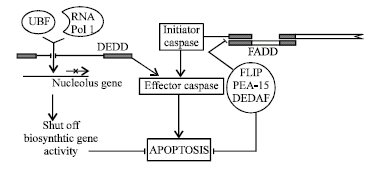 | |
| Fig. 4: | Role of different DED containing proteins in regulation of apoptosis |
CARD proteins, such as Apaf-1, Nod-1, Nod-2, DEFCAP, CARD12, IPAF, CLAN etc.
Coiled coil card: These CARD proteins are similar to NBD-CARD structurally, but lack NBD domain. Instead of NBD, coiled coil CARD consist of coiled coil dimerization motifs that act as an oligomerization surface. This group includes CARD 9/11/14/10 etc. These all are involved in NF-κB activation.
Bipartite CARD: These proteins are quite simpler to above mentioned CARD proteins and contain CARD plus one additional domain. All CARD containing caspase (Caspase-1, 2, 4, 5, 9) belong to this category. Apart from it, Rick, CARDIAK, RIP-2, Bcl-10, CIPER, mE10, RAIDD, ASC, CARDINAL, TUCAN and CARD-8 belong to this group.
CARD only protein: As the name suggest, these all protein consist of only CARD domain. These protein work as a positive or negative regulator of above mentioned proteins. These proteins include ICEBERG, COP etc (Bouchier-Hayes and Martin, 2002).
The first identified CARD molecule was Apaf-1, which interact with procaspase-9 in presence of Cytochrome C to form APOPTOSOME by CARD domain. However Nod-1 can directly bind to procaspase-9, independent of cytochrome C to form apoptosome (Saleh et al., 1999; Inohara et al., 1998).
Another CARD containing molecule mE10 (Mammalian E10) can also interact with procaspase-9 and resulted in apoptosis (Yan et al., 1999). Interaction of Apaf-1 to procaspase-9 can be inhibited by another CARD containing protein TUCAN (Tumor up regulated CARD containing antagonist of caspase nine) resulted in inhibition of caspase-9 activation and apoptosis inhibition (Pathan et al., 2001) (Fig. 5).
CARD proteins also plays major roles in receptor mediated apoptosis, such as Fas induced apoptosis require adaptor protein FADD, which recruit caspase-8, but interaction of caspase-8 to FADD can be prevented by ARC (Apoptosis repressor with CARD) and this ARC also bind and inhibit caspase-2 and 8 directly (Koseki et al., 1998).
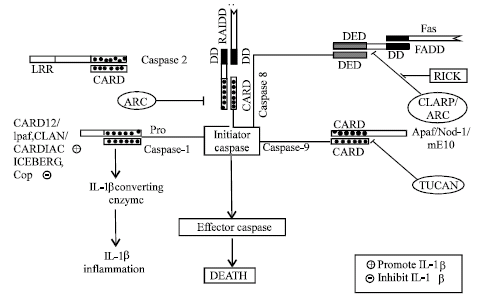 | |
| Fig. 5: | Role of CARD dimain containing proteins in regulation of apoptosis and IL-1β activity. Some CARD proteins induce apoptosis wile some inhibit in, see text for details |
Apoptosis mediated by interaction of caspase-8 to FADD can also be affected by caspase like molecule CLARP. But interaction of a CARD protein Rick to the CLARP resulted in Fas mediated apoptosis (Inohara et al., 1998). Fas mediated apoptosis can also be induced by CARD containing adaptor protein RAIDD. This adaptor molecule consists of DD in combination with CARD. RAIDD interact to Fas by DD and initiator caspase by CARD and resulted in apoptosis.
Another CARD containing protein DEFCAP interact with caspase-2 and caspase-9, DEFCAP consist of CARD, NBD, Leucine Rich Repeat (LRR). DEFCAP interact with CARD containing caspase by homotypic interaction and resulted in apoptosis induction in presence of LRR (Hlaing et al., 2001). Although CARD proteins play major roles in apoptosis, but these proteins are also involved in generation of cytokine IL-1β from pro IL-1β by IL-1β converting enzyme (ICE/caspase-1), a major regulator of inflammation. An Apaf like molecule Ipaf associate with procaspase-1 by CARD-CARD interaction and activate caspase-1 (Poyet et al., 2001). CARD12 and CLAN are also CARD family proteins that interact with caspase-1 and results in cytokine processing. But CARD12 is unusual, because it is involved in both cytokine processing and apoptosis (Geddes et al., 2001; Damiano et al., 2001). CARDIAK (CARD containing ICE associated kinase) is another protein, which interact with caspase-1 for generation of pro-inflammatory cytokine IL-1β. CARDIAK has also shown its involvement in NF-kB, Jun-N-terminal kinase activation (Thome et al., 1998). CARD-8, ICEBERG and COP (CARD only protein) also interact with caspase-1 but inhibit generation of IL-1β (Razmara et al., 2002; Humke et al., 2000; Lee et al., 2001). Apart form its role in apoptosis and inflammation, CARD proteins are major regulator of NF-κB activation (Fig. 6), CARDIAK, RIP-2, both interact with TRAF (TNFR associated factor), associated with TNFR, TRADD and RIP complex resulted in activation of IKK complex and NF-κB activation (McCarthy et al., 1998). cE10 (Another CARD protein) interact with TRAF related to DR4, TRADD and RIP complex and activate IKK complex, which in turn activate NF-κB (Costanzo et al., 1999). Some CARD acts downstream of these receptors, such as CARDINAL interact to IKKγ and CIPER interact with IKKβ, resulted in activation of NF-κB (Bouchier-Hayes et al., 2001; Koseki et al., 1999).
IAP (Inhibitor of apoptosis protein): IAP is characterized by presence of a 70 amino acid long domain called BIR (Baculovirus IAP repeat). Till now, various member of this IAP family have been identified, but not all are tested for their efficacy to suppress cell death. In addition to the BIR domain, IAP possess RING domain located near their carboxyl termini, such as xIAP, cIAP1, cIAP2 and drosophila IAP (DIAP1, DIAP2) etc. With the help of BIR domain, IAP protein thought to interact with various pro-apoptotic protein, resulted in their inactivation and inhibition of apoptosis (Fig. 7). Apart from their so called BIR, RING domain cIAP1 and cIAP2 also have CARD domain through which they interact with other CARD containing pro-apoptotic molecules (Devaraux and Reed, 1999).
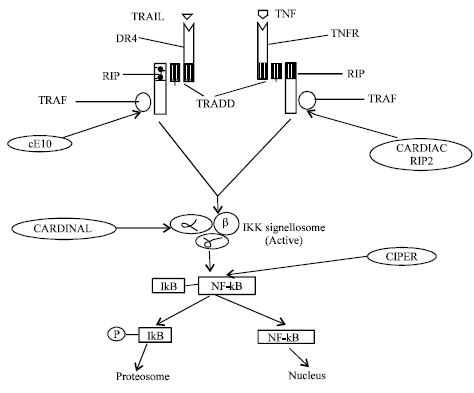 | |
| Fig. 6: | Role of CARD proteins in regulation of NF-kB activity. NF-kB plays major roles in regulation of apoptosis and inflammatory response |
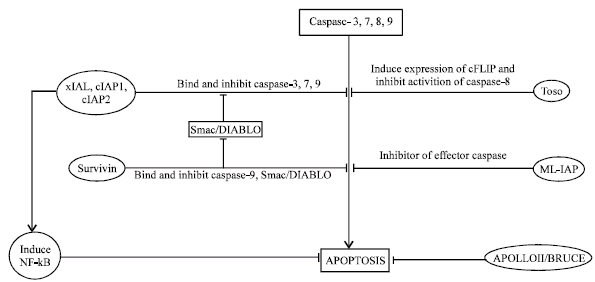 | |
| Fig. 7: | Roles of different inhibitor of apoptosis proteins. These carry IAP domain and are involved I regulation of apoptosis |
Some IAP protein such as Smac and its murine homologue DIABLO bind to other IAP proteins and remove their apoptosis inhibitory activity (Du et al., 2000; Verhagen et al., 2000). xIAP bind to caspase-3,7,9 and inhibit its activation (Deveraux et al., 1997). xIAP bind to caspase-3,7 due to interaction between Leu141 and Val 146 of xIAP and a hydrophobic site present on caspase-3,7. These Leu and Val are present in linker segment present within BIR2 domain of xIAP. XIAP bind to caspase-9 through the tetra peptide motifs present within caspase-9 and its interaction with BIR3 domain of xIAP, while Smac bind to xIAP and lead to formation of Smac-BIR3 and Smac-BIR2 complex, which resulted in activation of caspase-9 and caspase-3, 7, respectively (Fesik and Shi, 2001).
Another protein termed cIAP1 and cIAP2 also consist of three BIR domain and RING domain, through which they interact with caspase-3, 7, 9 and lead to subsequent ubiquitination and degradation in proteosome with the help of RING domain (Roy et al., 1997). cIAP1 and cIAP2 are also implicated in TNFR2-TRAF complex signaling. These protein bind to TRAF 1 and 2, through their BIR domain, this interaction suggest another mechanism of apoptosis suppression (Rothe et al., 1995). However, it has also been demonstrated that cIAP2 level rises in the cell with NF-κB induction (Chu et al., 1997) which is also involved with TNFR2-TRAF signaling.
Another IAP protein consisting single BIR and RING domain expressed in majority of melanoma cells. This protein called as ML-IAP (Melanoma IAP). It inhibits apoptosis in melanocytes and leads to melanoma (Skin cancer) (Vucic et al., 2000). NAIP (Neuronal apoptosis inhibitory protein) that inhibit apoptosis in neurons, is also a member of IAP family (Liston and Cor, 1996). NAIP has been shown to inhibit caspase 3,7. Survivin is also a prototype of IAP family and inhibit Caspase-9, Smac/DIABLO activation. It is expressed in various tumor cells but not in normal cells, so it acts as a diagnostic and therapeutic target in cancer cells (Pizem et al., 2003). Similarly, human IAP family protein Apollon and it homologue BRUCE are expressed in human cancer cells, where it inhibit apoptosis (Chen and Cor, 1999). Toso is another IAP protein, which activate cFLIP (discussed in DED) and inhibit caspase-8 processing and apoptosis (Hitoshi et al., 1998).
Bcl-2: The most diverse apoptosis regulating proteins comprised of B-cell lymphocytic leukemia proto-oncogene-2 (Bcl-2) family. Bcl-2 family is characterized by Bcl-2 homology (BH) domain. This family consist of both anti and pro apoptotic members of bcl-2 family. This family is divided into three groups on the basis of number of BH domain in each group. First group carry four BH domains (BH1-BH4) and all the member of this group are anti apoptotic, some examples of this group are Bcl-2, Bcl-XL, Bcl-w and Mcl-1. Next group carry only three BH domains and these all are involved in promotion of apoptosis. This group carries Bax, Bak and Bok. While next group is slightly far from above mentioned proteins because it carries only a single BH domain and these proteins are also involved in promotion of apoptosis. Examples of this group are Bik, Bim, Hrk/DP5, Noxa, Bad, Puma, Bmf, Bid etc. Apart from this so called BH domain, some bcl-2 family protein also carries a trans-membrane domain (TM), through which they interact with intracellular membrane of organelles. Such as, bcl-2 binds to nuclear envelope, E.R., outer mitochondrial membrane through its trans-membrane domain. Bcl-XL and Bcl-W localize mostly on outer mitochondrial membrane. While some proteins carries TM domains but remain cytosolic, because their hydrophobic c-terminal helix occlude their domains and flip out themselves to interact with intracellular membranes, so their activation requires displacement of c-terminal helix (Cory et al., 2003). As the structural information told us, that an anti-apoptotic Bcl-2 family member should have all four BH domain. It has been proposed, that BH4 domain interact with certain proteins involved in induction of apoptosis and inhibit their activity, such as BH4 domain interact with Ca++ dependent phosphatase calcineurin, so this calcineurin translocates to intracellular organelles membrane and this translocation of calcineurin prevent its activity on various cytosolic substrates. Such as phasphorylated NF-AT and this NF-AT is required for proliferation of some type of lymphocytes (Reed, 1997). However, this proposal is not true for all anti apoptotic bcl-2 family proteins. Actually, relative concentration of anti and pro apoptotic bcl-2 family member determines fate of the cell.
The precise mechanisms by which these proteins regulate apoptosis depend mostly on mitochondria and regulate mitochondrial membrane permeability. Due to defect in mitochondrial membrane permeability, mitochondria release various apoptosis stimulating substances. Now it has been postulated that, diverse apoptotic signal first converge on different BH-3 only domain, which deliver apoptotic signal to mitochondria by other members of the bcl-2 family proteins. This mitochondrial membrane permeabilization is achieved by creating pores in mitochondrial membrane by bcl-2 family proteins. Such as, BAX and Bak oligomerize to form pores inside mitochondrial membrane. However Bcl-2 inhibits apoptosis by inhibiting oligomerization of Bax or Bak and also by sequestering BH3 only proteins. So on apoptotic stimulation BH3 only protein inhibit activity of anti-apoptotic Bcl-2 family proteins Bcl-2, Bcl-XL etc. and induce BAX or BAK to form pores inside mitochondrial membrane and ultimately releases Cytochrome C, Endonuclease G, AIF to initiate intrinsic apoptosis pathway (Breckenridge and Xue, 2004). Although, Bcl-XL has also been shown to bind Apaf-1 and inhibiting its activity to form APOPTOSOME and BH3 only protein Bik interacts with Bcl-XL, upon apoptotic stimulation and neutralize its activity, resulted in formation of apoptosome by free Apaf-1 and procaspase-9, but most experimental evidences focuses towards pore formation by Bcl-2 family member in apoptosis (Adams and Cory, 1998). Another Bcl-2 family protein Bid unique in this respect, that it act as a connective link between extrinsic and intrinsic apoptosis pathways. In normal cells Bid is a cytosolic protein, but upon apoptotic stimulation Bid cleaved by caspase-8 through receptor mediated pathway and ultimately form tBid (truncated Bid). This tBid migrate to mitochondria to induce release of cytochrome C. tBid interact with an unusual lipid present in mitochondria (Cardiolipin). This cardiolipin help in migration - April 24, 2010tBid in mitochondria and release of cytochrome C (Degli-Esposti, 2004).
These all the proteins interact with each other and these signaling events result in a typical morphology of apoptosis. Caspase cleave cytoskeleton and cause cell shrinkage and ultimate generation of apoptotic bodies. These apoptotic bodies exhibit a signal to eat themselves that summon the scavenger cells. However these all signals are remain to be known, but it has been verified that phospholipid Phosphotidyl Serine (PS) act as a eat me signal. Normally PS is localized at the inner layer of plasma membrane bilayer, but in apoptotic bodies PS is exposed on the surface. PS interact with PS receptor present on macrophage. PS receptor is involved in engulfment of apoptotic bodies, as well as it signals a macrophage to reorganize its cytoskeleton, so it can surround and swallow apoptotic bodies. In some instances, complement cascade protein C1q also act as a receptor for its interaction with macrophage (Savill et al., 2003).
In conclusion, apoptosis is a major mechanism that initiates life in terms of development of embryo by giving it proper shape. This mechanism is imperative for maintenance of cellular homeostasis. The regulation is also crucial for an organism in order to sustain on its own. Besides that, if this balance of apoptosis and cell generation will disrupt, this can lead to various anomalies in the organism. Proper understanding of apoptotic pathways is helpful in unraveling the possible ways for treatment of cancer. Moreover, certain infectious agents including bacteria and virus induce changes in host cell to inhibit apoptosis and leads to development of cancer (Khan and Shrivastava, 2010). So, understanding of these pathways is helpful in counteracting such anomalies. As well as, this understanding is also helpful in finding out the mechanism of viral survival in a cell for a longer time, because virus can efficiently control these pathways in order to sustain their own. However this field is drawing attention of scientific community due to its interesting mechanism and scope in the medical field. Proper understanding of this intracellular mechanism is also very helpful for the treatment of many disease caused by viruses and bacteria. Scientists working on these intracellular mechanisms are very enthusiastic, that their effort will usher a new era in which they definitely will solve the problems of various drastic diseases like AIDS and Cancer by unraveling total mechanism of apoptosis. Apoptotic resistance is a major problem in front of scientific community while studying cancer. The cells become resistant to apoptosis even under the stimulus of several apoptotic signals. Proper understanding of this mechanism indeed can help in development of more realistic cancer control mechanisms. AIDS virus induce cells to initiate apoptosis, this reverse mechanism can be solved by understanding apoptotic resistance mechanisms. Control of this mechanism can give us control measure for several diseases.
ACKNOWLEDGMENT
AAK is thankful Dr. Mohit Maurya for critical reading and valuable suggestions during preparation of this manuscript.
REFERENCES
- Ahmed, M., S.M. Srinivasula, L. Wang, R.V. Talanian, G. Litwack, T. Fernandes-Alnemri and E.S. Alnemri, 1997. CRADD, a novel human apoptotic adaptor molecule for caspase-2 and FasL/tumor necrosis factor receptor-interacting protein RIP. Cancer Res., 57: 615-619.
PubMed - Barisic, K., J. Petric and L. Rumora, 2003. Biochemistry of apoptotic cell death. Acta Pharma., 53: 151-164.
Direct Link - Basu, A. and A. Miura, 2002. Differential regulation of extrinsic and intrinsic cell death pathways by protein kinase C. Int. J. Mol. Med., 10: 541-545.
PubMed - Beutler, B. and F. Bazzani, 1998. TNF, apoptosis and autoimmunity: A common thread. BCMD., 24: 216-230.
PubMed - Bhatnagar, P., A.A. Khan, M. Jain, S.K. Jain and A. Shrivastav, 2007. Bacteriological study of khoa sold in gwalior and morena city (Madhya Pradesh) in relation to public health. Asian J. Exp. Sci., 21: 55-62.
Direct Link - Bhatnagar, P., A.A. Khan, M. Jain S. Kaushik and S.K. Jain, 2007. Microbiological study of khoa sold in chambal region (Madhya Pradesh): A case study. Indian J. Microbiol., 47: 263-266.
CrossRefPubMedDirect Link - Bouchier-Hayes, L., H. Conroy, H. Egan, C. Adrain, E.M. Creagh, M. MacFarlane and S.J. Martin, 2001. CARDINAL, a novel caspase recruitment domain protein, is an inhibitor of multiple NF-kappa B activation pathways. J. Biol. Chem., 276: 44069-44077.
PubMed - Breckenridge, D.G. and D. Xue, 2004. Regulation of mitochondrial membrane permeabilization by BCL-2 family proteins and caspases. Curr. Opin. Cell Biol., 16: 647-652.
PubMedDirect Link - Castedo, M., J.L. Perfettini, T. Roumier, K. Andreau, R. Medema and G. Kroemer, 2004. Cell death by mitotic catastrophe: A molecular definition. Oncogene, 23: 2825-2837.
CrossRefPubMedDirect Link - Chen, Z., M. Naito, S. Hori, T. Mashima, T. Yamori and T. Tsuruo, 1999. A human IAP-family gene, apollon, expressed in human brain cancer cells. Biochem. Biophys. Res. Commun., 264: 264-854.
CrossRef - Chinnaiyan, A.M., K. O'Rourke, G. Yu, R.H. Lyons and M. Garg et al., 1996. Signal transduction by DR3, a Death Domain- containing receptor to TNFR-1 and CD95. Science, 274: 990-992.
PubMed - Chu, Z.L., T.A. Mckinsey, L. Liu, J.J. Gentry, M.H. Malim and D.W. Ballard, 1997. Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-kappaB control. Proc. Nat. Acad. Sci., 94: 10057-10062.
PubMed - Cory, S., D.C. Huang and J.M. Adams, 2003. The Bcl-2 family: Roles in cell survival and oncogenesis. Oncogene, 22: 8590-8607.
CrossRef - Costanzo, A., C. Guiet and P. Vito, 1999. c-E10 is a caspase-recruiting domain-containing protein that interacts with components of death receptors signaling pathway and activates nuclear factor-kappa B. J. Biol. Chem., 274: 20127-20132.
PubMed - Damiano, J.S., C. Stehlik, F. Pio, A. Godzik and J.C. Reed, 2001. CLAN, a novel human CED-4-like gene. Genomics, 75: 77-83.
PubMed - Deveraux, Q.L. and J.C. Reed, 1999. IAP family proteins-suppressors of apoptosis. Genes Dev., 13: 239-252.
PubMedDirect Link - Deveraux, Q.L., R. Takahashi, G.S. Salvesen and J.C. Reed, 1997. X-linked IAP is a direct inhibitor of cell-death proteases. Nature, 388: 300-304.
PubMed - Ding, W.X. and X.M. Yin, 2004. Dissection of multiple mechanisms of TNF-α induced apoptosis in liver injury. J. Cell Mol. Med., 8: 445-454.
CrossRef - Du, C., M. Fang, Y. Li, L. Li and X. Wang, 2000. Smac, a mitochondrial protein that promotes cytochrome C-dependent caspase activation by eliminating IAP inhibition. Cell, 102: 33-42.
PubMedDirect Link - Elzey, B.D., T.S. Griffith, D.M. Herndon, R. Barreiro, J. Tschopp and T.A. Ferguson, 2001. Regulation of Fas ligand-induced apoptosis by TNF. J. Immunol., 167: 3049-3056.
PubMed - Degli-Esposti, M., 2004. Mitochondria in apoptosis: Past, present and future. Biochem. Soc. Trans., 32: 493-495.
PubMed - Fesik, S.W. and Y. Shi, 2001. Controlling the caspases. Science, 294: 1477-1478.
CrossRefPubMedDirect Link - Geddes, B.J., L. Wang, W.J. Huang, M. Lavellee and G.A. Manji et al., 2001. Human CARD12 is a novel CED4/Apaf-1 family member that induces apoptosis. Biochem. Biophys. Res. Commun., 284: 77-82.
PubMed - Green, D.R. and J.C. Reed, 1998. Mitochondria and apoptosis. Science, 281: 1309-1312.
CrossRefPubMedDirect Link - Gupta, S., 2003. Molecular signaling in death receptor and mitochondrial pathways of apoptosis. Int. J. Oncol., 22: 15-20.
PubMed - Bouchier-Hayes, L. and S.J. Martin, 2002. CARD games in apoptosis and immunity. EMBO Rep., 3: 616-621.
CrossRef - Hitoshi, Y., J. Lorens, S.I. Kitada, J. Fisher and M. LaBarge et al., 1998. Toso, a cell surface, specific regulator of Fas-induced apoptosis in T cell. Immunity, 8: 461-471.
PubMed - Hlaing, T., R.F. Guo, K.A. Dilley, J.M. Loussia and T.A. Morrish et al., 2001. Molecular cloning and characterization of DEFCAP-L and -S, two isoforms of a novel member of the mammalian Ced-4 family of apoptosis proteins. J. Biol. Chem., 276: 9230-9238.
CrossRef - Hsu, H., J. Xiong and D.V. Goeddel, 1995. The TNF receptor 1-associated. protein TRADD signals cell death and NF-kappa B activation. Cell, 81: 495-504.
PubMed - Humke, E.W., S.K. Shriver, M.A. Starovasnik, W.J. Fairbrother and V.M. Dixit, 2000. ICEBERG: A novel inhibitor of interleukin-1beta generation. Cell, 103: 99-111.
PubMed - Inohara, N., L. Delpeso, T. Koseki, S. Chen and G. Nunez, 1998. RICK, a novel protein kinase containing a caspase recruitment domain, interacts with CLARP and regulates CD95-mediated apoptosis. J. Biol. Chem., 273: 12296-12300.
PubMed - Inohara, N., T. Koseki, L. Peso, Y. Hu and C. Yee et al., 1999. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappa B. J. Biol. Chem., 274: 14560-14567.
PubMed - Jiang, Y., J.D. Woronicz, W. Liu and D.V. Goeddel, 1996. Prevention of constitutive TNF receptor I signaling by silencer of death domains. Science 283: 543-546.
PubMed - Khan, A.A. and A. Shrivastava, 2010. Bacterial infection associated with cancer: possible implication in etiology with special reference to lateral gene transfer. Cancer Metastasis Rev.
CrossRef - Koseki, T., N. Inohara, S. Chen and G. Nunez, 1998. ARC, an inhibitor of apoptosis expressed in skeletal muscle and heart that interacts selectively with caspases. Proc. Nat. Acad. Sci., 95: 5156-5160.
PubMed - Koseki, T., N. Inohara, S. Chen, R. Carrio and J. Merino et al., 1999. CIPER, a novel NF kappaB-activating protein containing a caspase recruitment domain with homology to Herpesvirus-2 protein E10. J. Biol. Chem., 274: 9955-9961.
PubMed - Lee, S.H., C. Stehlik and J.C. Reed, 2001. COP, a caspase recruitment domain-containing protein and inhibitor of caspase-1 activation processing. J. Biol. Chem., 276: 34495-34500.
CrossRef - Li, P., D. Nijhawan, I. Budihardjo, S.M. Srinivasula, M. Ahmad, E.S. Alnemri and X. Wang, 1997. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell, 14: 479-489.
CrossRefPubMedDirect Link - Liston, P., N. Roy, K. Tamai, C. Lefebvre and S. Baird et al., 1996. Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature, 379: 349-353.
PubMed - McCarthy, J.V., J. Ni and V.M. Dixit, 1998. RIP2 is a novel NF-kappaB-activating and cell death-inducing kinase. J. Biol. Chem., 273: 16968-16975.
PubMed - Micheau, O. and J. Tschopp, 2003. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell, 114: 181-190.
CrossRefPubMedDirect Link - Muzio, M., B.R. Stockwel, H.R. Stennicke, V.S. Salvesen and V.M. Dixit, 1997. An induced proximity model for caspase-8 activation. J. Biol. Chem., 273: 2926-2930.
CrossRef - Nunez, G., M.A. Benedict, Y. Hu and N. Inohara, 1998. Caspases: The proteases of the apoptotic pathway. Oncogene, 17: 3237-3245.
PubMedDirect Link - Pan, G., J.H. Bauer, V. Haridas, S. Wang and D. Liu et al., 1998. Identification and functional characterization of DR6, a novel death domain-containing TNF receptor. FEBS Lett., 431: 351-356.
PubMed - Pathan, N., H. Marusawa, M. Krajewska, S. Matsuzawa and H. Kim et al., 2001. TUCAN, an antiapoptotic caspase-associated recruitment domain family protein overexpressed in cancer. J. Biol. Chem., 276: 32220-32229.
CrossRef - Peter, M.E., A.E. Heufelder and M.O. Hengartner, 1997. Advances in apoptosis research. Proc. Nat. Acad. Sci., 94: 12736-12737.
PubMed - Poyet, J.L., S.M. Srinivasula, M. Tnani, M. Razmara, T. Fernandes-Alnameri and E.S. Alnameri, 2001. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J. Biol. Chem., 276: 28309-28313.
PubMed - Ramos, J.W., P.E. Hughes, M.W. Renshaw, M.A. Schwartz, E. Formstecher, H. Chneiweiss and M.H. Ginsberg, 2000. Death effector domain protein pea-15 potentiates kinase by an adhesion-independent mechanism. Mol. Biol. Cell, 11: 2863-2872.
Direct Link - Razmara, M., S.M. Srinivasula, L. Wang, J.L. Poyet and B.J. Geddes et al., 2002. CARD-8 protein, a new CARD family member that regulates caspase-1 activation and apoptosis. J. Biol. Chem., 277: 13952-13958.
PubMed - Riedl, S.J. and Y. Shi, 2004. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol., 5: 897-907.
CrossRefPubMedDirect Link - Rothe, M., M.G. Pan, W.J. Henzel, T.M. Ayres and D.V. Goeddel, 1995. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell, 83: 1243-1252.
PubMed - Roy, N., Q.L. Deveraux, R. Takahashi, G.S. Salvesen and J.C. Reed, 1997. The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO. J., 16: 6914-6925.
CrossRef - Saleh, A., S.M. Srinivasula, S. Acharya, R. Fishel and E.S. Alnemri, 1999. Cytochrome c and dATP-mediated oligomerization of Apaf-1 is a prerequisite for procaspase-9 activation. J. Biol. Chem., 274: 17941-17945.
PubMed - Savill, J., C. Gregory and C. Haslett, 2003. Cell biology. Eat me or die. Science, 302: 1516-1517.
PubMed - Sharif-Askari, E., A. Alam, M. Rheaume, P.J. Beresford and C. Scotto et al., 2001. Direct cleavage of the human DNA fragmentation factor-45 by granzyme B induces caspase-activated DNase release and DNA fragmentation. EMBO. J., 20: 3101-3113.
PubMed - Sheshagiri, S. and L.K. Miller, 1997. Caenorhabditis elegans CED-4 stimulates CED-3 processing and CED-3-induced apoptosis. Curr. Biol., 7: 455-460.
PubMed - Siegel, R.M., D.A. Martin, L. Zheng, S.Y. Ng, J. Bertin, J. Cohen and M.J. Lenardo, 1998. Death-effector filaments: Novel cytoplasmic structures that recruit caspases and trigger apoptosis. J. Cell Biol., 141: 1243-1253.
CrossRef - Sprandio, S., I. Belle and D.E. Bredesen, 2000. An alternative, nonapoptotic form of programmed cell death. Proc. Nat. Acad. Sci., 97: 14376-14381.
PubMed - Stegh, A.H., O. Schickling, A. Ehret, C. Scaffidi and P. Christoph et al., 1998. DEDD, a novel death effector domain-containing apoptosis-inducing protein targeted to nucleoli. EMBO. J., 17: 5974-5986.
CrossRef - Sun, X., J. Yin, M.A. Starovasnik, W.J. Fairbrother and V.M. Dixit, 2002. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J. Biol. Chem., 277: 9505-9511.
CrossRef - Thome, M., K. Hoffmann, K. Burns, F. Martinon, J.L. Bodmer, C. Mattmann and J. Tschopp, 1998. Identification of CARDIAK, a RIP-like kinase that associates with caspase-1. Curr. Biol., 8: 885-888.
CrossRef - Thornberry, N.A. and Y. Lazebnik, 1998. Caspases: Enemies within. Science, 281: 1312-1316.
CrossRefPubMedDirect Link - Tibbetts, M.D., L. Zheng and M.J. Lenardo, 2003. The death effector domain protein family: Regulators of cellular homeostasis. Nature Immunol. 4: 404-409.
PubMed - Tschopp, J., M. Irmler and M. Thome, 1998. Inhibition of Fas death signals by FLIPs. Curr. Opin. Immunol., 10: 552-558.
PubMed - Vaux, D.L. and A. Strasser, 1996. The molecular biology of apoptosis. Proc. Natl. Acad. Sci. USA., 93: 2239-2244.
Direct Link - Verhagen, A.M., P.G. Ekert, M. Pakusch, J. Silke and L.M. Connolly et al., 2000. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell, 102: 43-53.
CrossRefPubMedDirect Link - Vicencio, J.M., L. Galluzzi, N. Tajeddine, C. Ortiz and A. Criollo et al., 2008. Senescence, apoptosis or autophagy. Gerontology, 54: 92-99.
CrossRef - Vucic, D., H.R. Stennicke, M.T. Pisabarro, G.S. Salvesen and V.M. Dixit, 2000. ML-IAP, a novel inhibitor of apoptosis that is preferentially expressed in human melanomas. Curr. Biol., 10: 1359-1366.
PubMed - Wang, X., 2001. The expanding role of mitochondria in apoptosis. Genes Dev., 15: 2922-2933.
PubMedDirect Link - Weber, C.H. and C. Vincenz, 2001. The death domain superfamily: A tale of two interfaces?. Trends Biochem. Sci., 26: 475-481.
PubMed - Yan, M., J. Lee, S. Schilbach, A. Goddard and V. Dixit, 1999. mE10, a novel caspase recruitment domain-containing proapoptotic molecule. J. Biol. Chem., 274: 10287-10292.
PubMed - Yanagisawa, J., M. Takahashi, H. Kanki, H. Yano-Yanagisawa and T. Tazunoki et al., 1997. The molecular interaction of fas and FAP-1. A tri-peptide blocker of human fas interaction with FAP-1 promotes Fas induced apoptosis. J. Biol. Chem., 272: 8539-8545.
PubMed - Zhang, J., D. Cado, A. Chen, N.H. Kabra and A. Winoto, 1998. Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature, 392: 296-300.
PubMed - Zheng, L., O. Schickling, M.E. Peter and M.J. Lenardo, 2001. The death effector domain-associated factor plays distinct regulatory roles in the nucleus and cytoplasm. J. Biol. Chem., 276: 31945-31952.
CrossRef