
A.A.A. Hamid
Department of Industrial Biotechnology, Faculty of Biosciences and Bioengineering, Universiti Technologi Malaysia, 81310 Skudai, Johor, Malaysia
S. Hamdan
Faculty of Science and Technology, School of Biosciences and Biotechnology, Universiti Kebangsaan Malaysia, Selangor, Malaysia
S.H.Z. Ariffin
Faculty of Science and Technology, School of Biosciences and Biotechnology, Universiti Kebangsaan Malaysia, Selangor, Malaysia
F. Huyop
Department of Industrial Biotechnology, Faculty of Biosciences and Bioengineering, Universiti Technologi Malaysia, 81310 Skudai, Johor, Malaysia
Journal of Biological Sciences
Year: 2010 | Volume: 10 | Issue: 3 | Page No.: 190-199







ABSTRACT
A phylogenetic analysis of an unknown strain AZZ2 isolated from volcanic area Gunung Sibayak Indonesia was performed. Their phylogenetic relationships were analysed using MEGA4 software® to ascertain its evolutionary distance by reconstructing a phylogenetic tree of these organisms. The evolutionary history and bootstrap consensus tree were inferred using the Neighbor-Joining method from 500 replicates. The tree is drawn to scale, with branch lengths (next to the branches) in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the p-distance method and were in the units of the number of base substitutions per site. Based on the partial 16S rDNA sequence determination, the strain showed high sequence similarity to Citrobacter sp. strain JC73/SL7. AZZ2 gene was also compared among known dehalogenase producing bacteria 16S rDNA genes. The results suggested that AZZ2 was closely related to the Serratia marcescens HL1. On the basis of phylogenetic identification only, AZZ2 was subjected to grow on 2,2-dichloropropionate (2,2DCP). The results suggested that strain AZZ2 can degrade 2,2DCP as expected similar to the characteristic of strain HL1 that can grow on halogenated compound. From this study, there was a possibility to predict the phenotype of newly isolated bacteria. The present findings also show that the evolutionary relationships of 16S rDNA gene strain AZZ2 were illustrated by phylograms and both topology are not in good agreement and may suggest an uncertainty of the origin of dehalogenases in volcanic area.
PDF Abstract XML References Citation
How to cite this article
A.A.A. Hamid, S. Hamdan, S.H.Z. Ariffin and F. Huyop, 2010. Molecular Prediction of Dehalogenase Producing Microorganism using 16S rDNA Analysis of 2,2-dichloropropionate (Dalapon) Degrading Bacterium Isolated from Volcanic Soil. Journal of Biological Sciences, 10: 190-199.
DOI: 10.3923/jbs.2010.190.199
URL: https://scialert.net/abstract/?doi=jbs.2010.190.199
DOI: 10.3923/jbs.2010.190.199
URL: https://scialert.net/abstract/?doi=jbs.2010.190.199
INTRODUCTION
Naturally occurring carbon-halogen covalent bonds are found widely throughout the biosphere in plants and animals. The major group is chlorinated compounds followed by brominated compounds with fluorinated and iodinated compounds few in number (Suida and DeBernadis, 1973). The role of many of these compounds is suggested to inhibit the growth of competing species (for example production of antibiotics, tetracycline and chloramphenicol). However, it is the release of man-made compounds, that has raised awareness of environmental issues relating to halogenated compounds. Halogenated compounds are extensively used as herbicides, insecticides, fungicides, insulators and lubricants. Dalapon or 2,2-dichloropropionic acid is an example of herbicide that used to control specific annual and perennial grasses like Quackgrass, Bermuda grass or Johnson grass. It is a selective herbicide that kills only certain plants and sparing non-target types of vegetation (Ashton and Crafts, 1973).
Microorganisms capable of utilizing halogenated aliphatic hydrocarbons as sole sources of carbon and energy are widely distributed and a large number of them have been isolated (Hardman, 1991; Leisinger and Bader, 1993; Olaniran et al., 2001, 2004; Jing and Huyop, 2007, 2008; Ismail et al., 2008; Thasif et al., 2009). Hydrolytic dehalogenases represent the key position in the degradation of haloaliphatic compounds. These enzymes catalyze the cleavage of carbon-halogen bonds by nucleophilic substitution, replacing the halogen ion by a hydroxyl group derived from water.
The comparison of the topology of phylogenetic trees based on 16S rDNA and functional gene sequence is another possible way to investigate of microbial evolution of a special trait for a specific functional gene for example, prediction of whether a newly isolated organism can produce dehalogenase enzyme. Current study describes the identification of an unknown organism from volcanic soil area of Gunung Sibayak Indonesia, a small stratovolcano overlooking the town of Berastagi in Northern Sumatra. Based on 16S rDNA gene sequence, the identity of newly isolated microorganism was determined and subsequently subjected growth on 2,2-dichloropropionate (2,2DCP) minimal medium as sole source of carbon and energy.
MATERIALS AND METHODS
Cultivation: Soil sample was taken from volcanic area of Gunung Sibayak. All strains were cultivated aerobically at 30°C in Luria Bertani (LB) solid medium. Several isolated bacteria were identified using 16S rDNA analysis. Based on identification analysis, a single potential strain was cultured at 30°C on a rotary shaker in 250 mL flask containing 100 mL minimal medium. The liquid minimal medium was prepared as 10x concentration basal salts containing K2HPO4.3H2O (42.5 g L-1), NaH2PO4.2H20 (10.0 g L-1) and (NH4)2SO4 (20.0 g L-1). The trace metals salts solution was prepared at 10x concentrate that contained Nitriloacetic Acid (NTA) (1.0 g L-1), MgSO4.7H2O (2.0 g L-1), FeSO4.7H2O (120.0 mg L-1), MnSO4.4H2O (30.0 mg L-1), ZnSO4.7H20 (30.0 mg L-1) and CoCl2.6H2O (10.0 mg L-1) in distilled water (Hareland et al., 1975). Minimal media for growing bacteria contained 10 ml of 10x basal salts and 10 mL of 10x trace metal salts per 100 mL of distilled water and were autoclaved (121°C, for 15 min).
The carbon source of 2,2DCP was sterilized by filtration and was added to the autoclaved salts medium to a final concentration of 20 mM. The extent of growth determined by measuring the absorbance at A680nm and the release of chloride ions.
Molecular analysis: DNA was isolated from bacterial cells by using Wizard genomic purification kit (Promega). The 16S rDNA were amplified from purified DNA by PCR using Pfu DNA polymerase with the buffer supplied by the manufacturer (Promega). Universal 16S rDNA primers FD1 (5’-aga gtt tga tcc tgg ctc ag-3’) and rP1 (5’-acg gtc ata cct tgt tac gac tt-3’) (Fulton and Cooper, 2005) were synthesized by 1st BASE Laboratory Malaysia Sdn. Bhd. DNA sequencing was performed using ABI PRISM ® 377 DNA sequencer (1st BASE Laboratory Malaysia Sdn. Bhd).
Sequencing alignment and construction of phylogenetic tree: The 16S rDNA sequence obtained were aligned and compared with the sequences stored in Gene Bank from National Center for Biotechnology Information (NCBI) using BLASTn analysis tool. Multiple sequence alignment/phylogenetic tree was established using MEGA4 Molecular software.
Phylogenetic tree reconstruction using mega4 software: Phylograms of unknown bacteria were reconstructed using Mega4 Software (Tamura et al., 2007). Initially, many 16S rDNA gene sequences from different kinds of bacteria (related sequences/dehalogenase producing bacteria) that obtained from NCBI were added or pasted into alignment explorer/Clustal W by integrated web-browser. All sequence were aligned together to achieve multiple sequence alignment by clicking align by clustalW. After complete alignment by Clustal W, all output data were used together to reconstruct phylogram. The evolutionary history was inferred using the Neighbor-Joining method (Saitou and Nei, 1987). The bootstrap consensus tree inferred from 500 replicates is taken to represent the evolutionary history of the taxa analyzed. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) is shown next to the branches (Felsenstein, 1985). The tree is drawn to scale, with branch lengths (next to the branches) in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the p-distance method (Tamura et al., 2004) and are in the units of the number of base substitutions per site. All positions containing gaps and missing data were eliminated from the dataset (Complete deletion option).
RESULTS
Bacteria isolation: Several morphologically different colonies were observed on LB media after overnight growth at 30°C. Colonies formed were repeatedly streaked on the same type of medium in order to obtain a pure colony. Each pure colony was then subjected to 16S rDNA analysis.
Analysis of 16S rDNA gene sequence for genus identification: Genomic DNA of several bacteria species was prepared using Wizard Genomic DNA kit (Promega). The PCR amplification using appropriate primers revealed a single fragment of approximately 1.5 kb for each strains. The PCR products were purified using QIAquick PCR purification kit (Qiagen) for DNA sequencing. In order to get some idea concerning genera and species type, the nucleotide sequencing data were than analyzed using BLASTn online analysis tool to identify the closest phylogenetic relatives.
 | |
| Fig. 1: | The 16S rDNA partial sequence comparison of Citrobacter sp. JC73/SL7 with strain AZZ2 – showing 98% identity |
The gene sequences were compared to the sequences in the GenBank database-NCBI (US National Centre for Biotechnology Information).
Among all bacteria strains, AZZ2 showed the highest sequence similarity (98% sequence identity with e-value = 0) to Citrobacter sp. JC73/SL7 (Accession number: FN547926) (Fig. 1). Thus, it was designated as Citrobacter sp. strain AZZ2. In support to the current data, Gram staining suggested the species was a Gram negative, rod in shape.
Growth in 2,2DCP liquid minimal media: Based on the identification by 16S rDNA analysis, a single potential bacterial culture designated as strain AZZ2 was then tested to grow in liquid minimal medium supplied with 2,2DCP as sole source of carbon and energy. Strain AZZ2 was inoculated into 100 mL minimal liquid medium containing 20 mM 2,2DCP as the sole source of carbon. The flasks were incubated at 30°C in a rotary incubator at 180 rpm. The maximum growth was achieved after 48 h with cells doubling time 15 h.
Evolutionary relationship of Citrobacter sp. AZZ2: Phylogenetic tree was established using BLAST-Webpage (NCBI). According to Fig. 2, strain AZZ2 was located among Citrobacter sp. Further analysis was carried out by taking ten different related species of Citrobacter sp. as Operational Taxonomic Units (OTUs) (Tamura et al., 2004) in order to investigate the evolutionary relationship of Citrobacter sp. AZZ2 among related species (Table 1). There are 15047 base nucleotides of 16S rDNA gene sequences were analyzed (Table 2) and multiple alignment were constructed using Clustal W in MEGA4.
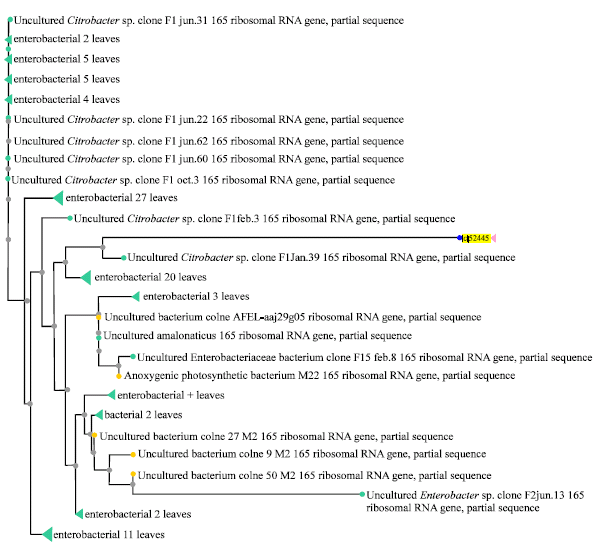 | |
| Fig. 2: | An overview of phylogenetic analysis of AZZ2 (marked as lcl 52445) using BLASTn-Webpage |
| Table 1: | Ten closest sequence of Citrobacter sp. AZZ2 selected from NCBI |
 | |
Numbers of base substitutions per site from pairwise distance analysis between sequences were shown in Fig. 3. All results are based on the pairwise analysis of 11 sequences. According to Fig. 3, the lowest value of genetic distance from Citrobacter sp. AZZ2 was 0.018 base substitutions per site. This value is due to the distance between Citrobacter sp. AZZ2 and Citrobacter sp. JC73/SL7. All pairwise distance analysis were conducted using the p-distance method in MEGA4. The proportion of observed distance, sometimes also called p-distance and it is expressed as the number of nucleotide differences site. All positions containing gaps and missing data were eliminated from the dataset (Complete deletion option).
| Table 2: | Nucleotide bases analysis among related Citrobacter sp. AZZ2 |
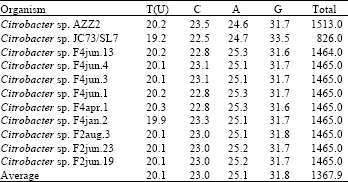 | |
There were a total of 826 positions in the final dataset. Values in Fig. 3 were programmed into Fig. 4 with optimal bootstrap consensus tree with the sum of branch length = 0.0236. Results strongly suggested that Citrobacter sp. AZZ2 was not located within the large different homologous sites is called clade of related species but it was closely related to the Citrobacter sp. JC73/SL7 with genetic distance 0.018 base substitutions per site.
 | |
| Fig. 3: | The number of base substitutions per site from analysis between sequences. All results are based on the pairwise analysis of 11 sequences. Analysis were conducted using the p-distance method in MEGA4. All positions containing gaps and missing data were eliminated from the dataset (Complete deletion option). There were a total of 826 positions in the final dataset |
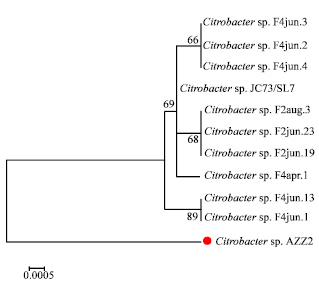 | |
| Fig. 4: | Phylogenetic relationship between Citrobacter sp. AZZ2 and other bacteria in same genera based on 16S rDNA sequences. The scale bar represents 0.0005 substitutions per site. Bootstrap values above 64% are shown at the nodes (based on 500 resamplings) |
| Table 3: | List of dehalogenase producing bacteria selected from NCBI |
 | |
Evolutionary relationship of Citrobacter sp. AZZ2 among dehalogenase producing bacteria: In this study, eleven dehalogenase producing bacteria and Citrobacter sp. AZZ2 were selected as Operational Taxonomic Units (OTUs) in order to investigate the evolutionary relationship of Citrobacter sp. AZZ2 among dehalogenase producing bacteria (Table 3). There are 15862 base nucleotides from 16S rDNA gene of Citrobacter sp. AZZ2 and related species were analyzed (Table 4) and multiple alignment were constructed using Clustal W in MEGA4.
 | |
| Fig. 5: | The number of base substitutions per site from analysis between sequences. All results are based on the pairwise analysis of 12 sequences. Analysis were conducted using the p-distance method in MEGA4. All positions containing gaps and missing data were eliminated from the dataset (Complete deletion option). There were a total of 486 positions in the final dataset |
| Table 4: | Base nucleotides analysis of Citrobacter sp. AZZ2 and dehalogenase producing bacteria |
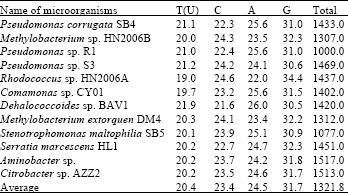 | |
Numbers of base substitutions per site from pairwise distance analysis between sequences were shown in Fig. 5. All results are based on the pairwise analysis of 12 sequences. According to the data in Fig. 5, the lowest value of genetic distance from Citrobacter sp. AZZ2 was 0.123 base substitutions per site. This value is due to the distance between Citrobacter sp. AZZ2 and Serratia marcescens HL1. All pairwise distance analysis were conducted using the p-distance method in MEGA4. All positions containing gaps and missing data were eliminated from the dataset (Complete deletion option). There were a total of 486 positions in the final dataset.
The evolutionary history was inferred using the Neighbor-Joining method. The bootstrap consensus tree inferred from 500 replicates is taken to represent the evolutionary history of the taxa analyzed. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) is shown next to the branches. The optimal tree with the sum of branch length = 0.912 is shown in Fig. 6. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. In the phylogram, there were twelve Operational Taxonomic Units (OTUs) which include Citrobacter sp. AZZ2 and eleven dehalogenase producing bacteria. This unrooted phylogram was inferred with ten internal nodes (hypothetical taxonomic units) represents ancestor of the OTUs. In Fig. 6, Citrobacter sp. AZZ2 was located within the same clade with Serratia marcescens HL1 with 100% of bootstrap value. Citrobacter sp. AZZ2 was most related with Serratia marcescens HL1 with genetic distance 0.123 base substitutions per site. Citrobacter sp. AZZ2 was having a distant relationship with Dehalococcoides sp.
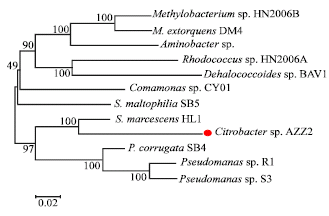 | |
| Fig. 6: | Phylogenetic relationship between Citrobacter sp. AZZ2 and dehalogenase producing bacteria 16S rDNA sequences. The scale bar represents 0.02 substitutions per site. Bootstrap values above 48% are shown at the nodes (based on 500 resamplings) |
BAV1 with genetic distance 0.307 base substitutions per site.
E. coli BL21 as positive and negative controls: The partial 16S rDNA gene sequence from E. coli BL21 was blasted in the NCBI database. The most significant result is shown in Fig. 7. From the results obtained, sequence from E. coli BL21 shared at least 97% identity to the entire top twenty most significant alignments. In negative control, the 16S rDNA gene sequence of E. coli BL21 was compared to Citrobacter sp. AZZ2. From 1049 base compared, 197 gaps were revealed and these two sequences shared 76.1% sequence identity (Fig. 8). This suggests that any identity percentage less than 76.1% is not significant in the identification of organism using 16S rDNA technique.
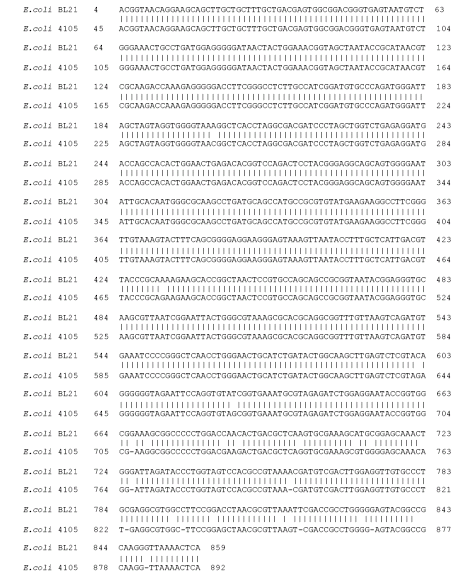 | |
| Fig. 7: | Sequence comparison of E. coli BL21 16S rDNA gene with E. coli 4105 showing 97% identity |
 | |
| Fig. 8: | Sequence comparison of E. coli BL21 16S rDNA gene with Citrobacter sp. AZZ2 showing 76.1% identity |
DISCUSSION
Bacteria taxonomy using 16S rDNA is a common method in the characterization and identification of microorganism, such as the taxonomy of actinomycetes (Colquhoun et al., 1998), taxonomy of extremophiles (Takami et al., 1997; Sorokin et al., 2000) and taxonomy of hydrocarbon degrading bacteria (Wang et al., 1995).
In current investigation, the 16S rDNA gene sequence from E. coli BL21 was included as positive and negative controls. The identity of E. coli agreed to the sequence in the database. For negative control, pairwise sequence alignment of the two sequences revealed sequence identity of 76.1%. This suggests, any identity less than 76.1% could not be accepted during identification of the organism, whereas sequence identity higher than 76.1% may be a significant value. These results for both controls strongly suggest the use of universal primers and DNA sequencing method could give a reliable identification of an unknown species.
BLASTn analysis revealed strain AZZ2 gene sequence shared at least 98% identity to the sequence of the Citrobacter sp. JC73/SL7 suggesting strain AZZ2 belongs to Citrobacter sp. In addition, this support the results obtained from Gram staining since Citrobacter sp. is a Gram negative, rod in shape. The genus Citrobacter had a potential to degrade halogenated compound by producing dehalogenase enzyme. Earlier study proven that this species has been isolated and could degrade chlorinated compound as carbon source. From Cluster analysis of RAPD pattern and respirometric data, the isolated bacteria was identified by using 16S rDNA sequence analysis as Citrobacter freundii strain HPC255. The strain HPC255 could oxidize different substituted chlorophenol molecules (Gurpreet et al., 2004).
In current study it was hypothesized that Citrobacter sp. AZZ2 is a dehalogenase producing bacteria strictly based on relatedness to the genus Citrobacter sp. and Serratia marcescencs HL1 (Li et al., 2008). These two types of bacteria could degrade halogenated compound as reported. According to the growth experiment, Citrobacter sp. AZZ2, could grow on 2,2DCP as sole source of carbon and energy. Therefore, it was possible to predict for a specific functional gene despite of to investigate microbial evolution of a special trait. Current strain deserved more studies since, there was no Citrobacter strain reported to grow in 2,2DCP minimal medium as a carbon source in the previous literature. This may shed light on isolating of thermostable dehalogenase from volcanic area.
In conclusion, a group bacteria isolated from soil was screened using 16S rDNA analysis. In the present investigation, we have further analyzed strain AZZ2. This organism was able to grow on 2,2DCP and had a closed evolutionary relationship with Citrobacter and Serratia. This was the first reported strain from Citrobacter that can degrade 2,2DCP. In future, it was hoped that such studies would be possible to isolate organism with desired characteristics.
ACKNOWLEDGMENTS
Authors would like to thank Ministry of Higher Education (MOHE-Vot 78307) for financial support and also Molecular Biology Laboratory (UKM) for DNA analysis.
REFERENCES
- Colquhoun, J.A., S.C. Heald, L. Li, J. Tamaoka, C. Kato, K. Horikoshi and A.T. Bull, 1998. Taxonomy and biotransformation activities for some deep-sea Actinomycetes. Extremophiles, 2: 269-277.
PubMed - Felsenstein, J., 1985. Confidence limits on phylogenies: An approach using the bootstrap. Evolution, 39: 783-791.
CrossRefDirect Link - Fulton, C.K. and R.A. Cooper, 2005. Catabolism of sulfamate by Mycobacterium sp. CF1. Environ. Microbiol., 7: 378-381.
Direct Link - Gurpreet, K.N., A. Kapley and H.J. Purohit, 2004. Isolation and characterization of Citrobacter strain HPC255 for broad-range substrate specificity for chlorophenols. Curr. Microbiol., 48: 419-423.
CrossRef - Hareland, W.A., R.L. Crawford, P.J. Chapman and S. Dagley, 1975. Metabolic function and properties of a 4-hydroxyphenylacetic acid 1-hydroxylase from Pseudomonas acidovorans. J. Bacteriol., 121: 272-285.
Direct Link - Hardman, D.J., 1991. Biotransformation of halogenated compounds. Crit. Rev. Biotechnol., 11: 1-40.
CrossRef - Ismail, S.N., A.M. Taha, N.H. Jing, R.A. Wahab, A.A. Hamid, R.V. Pakingking Jr. and F. Huyop, 2008. Biodegradation of monochloroacetic acid by a presumptive Pseudomonas sp. strain R1 bacterium isolated from Malaysian paddy (rice) field. Biotechnology, 7: 481-486.
CrossRefDirect Link - Jing, N.H. and F. Huyop, 2007. Dehalogenation of chlorinated aliphatic acid by Rhodococcus sp. Asia Pac. J. Mol. Biol. Biotechnol., 15: 147-151.
Direct Link - Jing, N.H. and F. Huyop, 2008. Enzymatic dehalogenation of 2,2-dichloropropionic acid by locally isolated Methylobacterium sp. HJ1. J. Biol. Sci., 8: 233-235.
CrossRefDirect Link - Leisinger, T. and R. Bader, 1993. Microbial dehalogenation of synthetic organohalogen compounds: Hydrolytic dehalogenases. Chimia, 47: 116-121.
Direct Link - Li, M.T., L.L. Hao, L.X. Sheng, J.B. Xu, 2008. Identification and degradation characterization of hexachlorobutadiene degrading strain Serratia marcescens HL1. Bioresour. Technol., 99: 6878-6884.
CrossRef - Olaniran, A.O., G.O. Babalola and A.I. Okoh, 2001. Aerobic dehalogenation potentials of four bacterial species isolated from soil and sewage sludge. Chemosphere, 45: 45-50.
CrossRef - Olaniran, A.O., D. Pillay and B. Pillay, 2004. Haloalkane and haloacid dehalogenases from aerobic bacterial isolates indigenous to contaminated sites in Africa demonstrate diverse substrate specificities. Chemosphere, 55: 27-33.
CrossRef - Saitou, N. and M. Nei, 1987. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol., 4: 406-425.
CrossRefPubMedDirect Link - Sorokin, D.Y., B.E. Jones, J.G. Kuenen, 2000. An obligate methylotrophic, methane-oxidizing Methylomicrobium species from a highly alkaline environment. Extremorphiles, 4: 145-155.
CrossRef - Suida, J.F. and J.F. DeBernadis, 1973. Naturally occuring halogenated organic compounds. Lloydia, 36: 107-143.
PubMed - Takami, H., A. Inoue, F. Fuji and K. Horikoshi, 1997. Microbial flora in the deepest sea mud of the Mariana Trench. FEMS Microbiol. Lett., 152: 279-285.
PubMed - Tamura, K., M. Nei and S. Kumar, 2004. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. U.S.A., 101: 11030-11035.
CrossRefDirect Link - Tamura, K., J. Dudley, M. Nei and S. Kumar, 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol., 24: 1596-1599.
CrossRefPubMedDirect Link - Thasif, S., S. Hamdan and F. Huyop, 2009. Degradation of D,L-2-chloropropionic acid by bacterial dehalogenases that shows stereospecificity and its partial enzymatic characteristics. Biotechnology, 8: 264-269.
CrossRefDirect Link - Wang, R.F., W.W. Cao and C.E. Carniglia, 1995. Phylogenetic analysis of polycyclic aromatic hydrocarbon degrading mycobacteria by 16S rRNA sequencing. FEMS Microbiol. Lett., 130: 75-80.
CrossRef