
Mohammad Mohiuddin
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Dhaka, Dhaka-1000, Bangladesh
A.T.M. Zafrul Azam
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Dhaka, Dhaka-1000, Bangladesh
Md. Shah Amran
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Dhaka, Dhaka-1000, Bangladesh
Md. Amjad Hossain
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Dhaka, Dhaka-1000, Bangladesh
Journal of Biological Sciences
Year: 2009 | Volume: 9 | Issue: 5 | Page No.: 476-481







ABSTRACT
The in vitro study of protein binding of caffeine and its 1:1 mixtures with gliclazide and metformin HCI has been conducted by equilibrium dialysis method performing measurement by direct spectrophotometric method at temperature 37± 0.5°C and at pH 7.4. In this study, number of binding sites and affinity constants of caffeine and its 1:1 mixture with gliclazide and metformin HCI were calculated by scatchard method. Scatchard plots were prepared to reveal the number of binding sites and the affinity for protein binding. It has been found that both gliclazide and metformin HCI cause lowering the affinity and percentage of binding of the drugs in the mixture to bovine serum albumin. Thus, the interaction of caffeine with gliclazide and metformin HCl can increase the free drug concentration of caffeine in blood plasma. This may change the pharmacokinetic and pharmacodynamic properties of the drug.
PDF Abstract XML References Citation
How to cite this article
Mohammad Mohiuddin, A.T.M. Zafrul Azam, Md. Shah Amran and Md. Amjad Hossain, 2009. In vitro Effects of Gliclazide and Metformin on the Protein Binding of Caffeine in the Aqueous Media. Journal of Biological Sciences, 9: 476-481.
DOI: 10.3923/jbs.2009.476.481
URL: https://scialert.net/abstract/?doi=jbs.2009.476.481
DOI: 10.3923/jbs.2009.476.481
URL: https://scialert.net/abstract/?doi=jbs.2009.476.481
INTRODUCTION
Caffeine is used as a mild CNS stimulant (Troy and Beringer, 2005). It is absorbed readily after oral administration and is widely distributed throughout the body. It is also absorbed through the skin. Caffeine passed readily into the CNS and into saliva; low concentrations are also present in breast milk (Sweetman, 2005). Caffeine crosses the placenta. In adults, caffeine is metabolized almost completely in the liver via, oxidation, demethylation and acetylation and is excreted in the urine and other metabolites with only about 1% unchanged. Neonates have a greatly reduced capacity to metabolize caffeine and it is largely excreted unchanged in the urine until hepatic metabolism becomes significantly developed, usually by about 6 months of age. Elimination half-lives are about 3 to 7 h in adults but may be in excess of 100 h in neonates (Sweetman, 2005). Gliclazide is a sulfonylurea antidiabetic. It is readily absorbed from the gastrointestinal tract. It is extensively bound to plasma proteins. The half-life is about 10 to 12 h. Gliclazide is extensively metabolized in the liver to metabolites that have no significant hypoglycemic activity. Metabolites and a small amount of unchanged drug are excreted in the urine (Kobayashi et al., 1984). Metformin hydrochloride is a biguanide antidiabetic. It is slowly and incompletely absorbed from the gastrointestinal tract; the absolute bioavailability of a single 500 mg dose is reported to be about 50-60%, although this is reduced somewhat if taken with food. Once absorbed plasma protein binding is negligible and it is excreted unchanged in the urine. The plasma elimination half-life is reported to range from about 2-6 h after oral doses. Metformin is distributed into breast milk in small amounts (Scheen, 1996; Sambol et al., 1996).
Protein binding is one of the important pharmacokinetic parameters of a drug. After oral administration the drug enters the systemic circulation through absorption and binds with plasma protein of blood. Among plasma proteins, albumin is highly bound to drugs. It contains of 585 amino acids, having molecular weight about 69000. Its concentration is also high in blood e.g., 3.4-4.5 g dL-1 (about 60% of total the circulating proteins). Other important plasma proteins are α-globulin and γ-globulin. The interaction of a drug with protein may be reversible or irreversible. In reversible case, the drug-protein complex acts as a reservoir and release the unbound (free) drug and equilibrium exists between bound and unbound fractions of a drug. Drugs are bound to plasma protein at sites located on the surface of the protein. There are some drugs, which bind to some specific sites in the protein molecule; those sites are then named according to the drugs, which bind to it. Generally three types of protein binding sites are observed. These are warfarin site: site I, diazepam site: site II and digoxin site: site III. The idea of binding sites is suggested by the relative sizes of the drugs and proteins. Drugs are small molecule with a molecular weight of the order of 150-400 and occupy only a small area of the large protein molecules (Singlas, 1987; Joseph, 1982).
The extent of plasma protein binding is an important parameter of drug action. Binding to plasma protein may have a profound effect on distribution, pharmacological action and rate of elimination. Distribution is a physicochemical interaction between a drug and the body and is governed by the two components involved in the interaction. The distribution of a drug from blood to other tissue fluids is measured by its apparent volume of distribution. A large volume of distribution indicates extensive tissue diffusion i.e., drug is distributed throughout the body. Conversely, a small volume of distribution is a sign of retention or low tissue uptake. Mathematically:
Where:
| D | : | Total amount of drug in the body |
| C0 | : | Plasma concentration of drug at zero time |
Kidney and liver are mostly responsible for drug elimination. In pharmacokinetics, the elimination of a drug, whether by renal or hepatic pathway is expressed by its clearance. In case of renal elimination, Glomerular Filtration (GFR) of a drug is carried out by passive mechanism and higher protein binding lowers the GFR of a drug. In case of hepatic elimination when the drug has higher affinity to plasma protein than hepatic cells, then protein lowers the hepatic elimination of the drug and the hepatic clearance does not depend on the hepatic blood flow. The free (unbound) drug concentration in plasma rather than the total plasma concentration (bound and unbound drug) determines the effect of drug. Simultaneous administration of two or more drugs can modify the affinity of the drug to plasma protein and thus percentage of protein binding. Due to this modification, the volume of distribution, renal and hepatic clearance of drug can be changed by combined therapy and drug effect can be modified by Singlas (1987) and Cadwallader (1985).
Drug-drug interaction result when one drug alters the known therapeutic response of another that has been administered concurrently or before or after the drug. The next result may be enhanced or diminished effects of one or both the drugs (Hansten and Horn, 1989). A common practice in the medical science is the prescription of multiple drugs at a time, which may sometimes be neither safe nor effective and may be deleterious. Over the last 10 year, the research on drug-drug interactions, drug-metal interactions and drug-food interactions were carried out by Bari et al. (2000) and Amran et al. (2006a, b, 2008). In present, continuous study on the fate of multiple drug use, we have studied the effects of gliclazide and metformin on the protein binding of caffeine in the aqueous medium.
This study was aimed to evaluate the influence of gliclazide and metformin hydrochloride on the percentage of protein binding of caffeine at physiological pH (7.4) and temperature (37± 0.5°C) and thus to infer about the combination therapy.
MATERIALS AND METHODS
Materials: Caffeine, gliclazide and metformin HCI kind gift from the Orion Laboratories Ltd., Dhaka, Bangladesh.
Reagents: Bovine serum albumin (FractionV, 96-98%, SIGMA), semi-permeable membrane (Mediciel, England), sodium bi-carbonate, M/15 phosphate buffer, hydrochloric acid (37%, reagent grade), potassium dihydrogen orthophosphate (reagent grade, Merck, Germany), disodium hydrogen orthophosphate (peagent grade, Merck, Germany), orthophospharic acid (reagent grade), potassium hydroxide (reagent grade), sodium hydroxide (reagent grade, Merck, Germany) and demineralized water (Orion Laboratories Ltd., Dhaka, Bangladesh).
The protein binding experiments were carried out according to the procedure of the earlier studies by Amran et al. (2008). The experimental methods are also discussed briefly herein.
Preparation of Bovine Serum Albumin (BSA) solution: One hundred milliliter solution of 5x10-5 M was prepared by dissolving 0.3450 g of bovine serum albumin (MW 6900 g) in M/15 phosphate buffer having pH 7.4 and make the volume up to 100 mL with the same solvent.
Equipments: UV-Visible Spectrometer (Model No. UV-1601, Shimadzu, Japan), pH Meter (Mettler Toledo, Switzerland), Power Sonic (Model No. 510, Seoul, Korea), Analytical Balance (Sartorious, Model No. BL-2105, Germany) and Dubnoff metabolic shaking incubator (GCA corporation, USA).
Preparation of standard solutions: Caffeine, gliclazide and metformin HCl were dissolved in demineralized water separately. These stock solutions were diluted to desired strengths by buffer solution to get the working standard solution (20 μg mL-1).
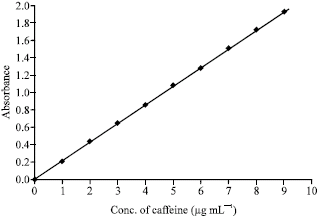 | |
| Fig. 1: | Standard curve of caffeine for equilibrium dialysis method |
Preparation of standard curve: For the spectrophotometric determination of drug concentration into the buffer compartment A, a standard curve was used. To prepare the standard curve, a treated M/15 phosphate buffer (pH 7.4) was used. Solution of different concentrations of caffeine were prepared in this buffer and a standard curve was prepared by plotting absorbance (measured at 273 nm) against concentrations (Fig. 1).
Equilibrium dialysis method: Equilibrium dialysis (Singlas, 1987) is one of the methods used for the determination of protein binding of any compound. This method was developed by Singlas (1987) which consists in dialyzing the unbound fraction of a compound contained in a protein (bovine serum albumin) solution through a semi-permeable membrane. In this study, this method was used for the determination of protein binding of caffeine and its 1:1 mixture with gliclazide and metformin HCl. In this method firstly, dialysis membrane was activated and then dialysis was performed.
Activation procedure: The dialysis membrane were cut into 12 cm pieces look like a bag (also called dialysis bag) and immersed in boiling 1 M NaHCO3 solution for about 1 h to make sure that the inside of the bag was washed as well as outside and the process was repeated once.
Then these bags were immersed in boiling demineralized water for about 1 h with intermittent change of the water making sure that all the anions and cations are washed out. Then these were well washed with demineralized water.
Then these were immersed in M/15 phosphate buffer having pH 7.4 (Perrin and Dempsey, 1974; Bates, 1964) at about 70°C for 1 h. The process was repeated once.
Finally, these were rinsed with demineralized water and stored in a refrigerator with the same buffer.
Dialysis procedure: The activated membrane were filled with Bovine Serum Albumin (BSA) solution with different concentrations of caffeine or its 1:1 mixture with gliclazide and metformin HCl, keeping the total volume 3 mL. Then, these were immersed in a fixed amount (25 mL) of M/15 phosphate buffer having pH 7.4 in a 100 mL conical flask.
Conical flasks were shaken gently at 37± 0.5°C for about 8 h in a Dubnoff metabolic shaking incubator. After completion of dialysis, the absorbance of buffer (outside the membrane) was measured at 273 nm using the UV-VIS recording spectrophotometer.
Calculation of percentage of protein binding: Initially, a known amount of drug was given into plasma compartment (dialysis bag). Then, concentration of drug present in the buffer (outside of this compartment) after equilibrium was measured. This measurement gave the total amount of drug that remains in the dialysis bag. Thus, we can get sum of free drug and plasma bound drug at equilibrium.
The percentage of protein binding (F) of the drug is calculated using the following equation,
Where:
| [A] | : | Molar conc. of drug in buffer compartment |
| [B] | : | Molar conc. of drug in plasma compartment which found by subtracting [A] from the initial conc. of the drug (amount of drug in plasma after equilibrium) |
Calculation of number of binding sites and the affinity constants: In the present study, number of binding sites and affinity constants of caffeine and its 1:1 mixture with gliclazide and metformin HCl were calculated by scatchard method (Singlas, 1987; Goldstein et al., 1974; Scatchard, 1949).
In this method, a curve was produced by plotting r/[D] versus r, where r is the ratio between the molar concentration of the bound drug and the molar concentration of protein i.e.,
and D is the concentration of the unbound drug i.e., [A].
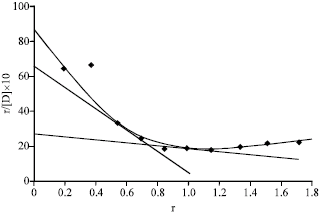
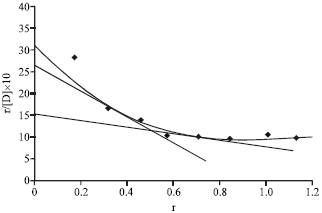
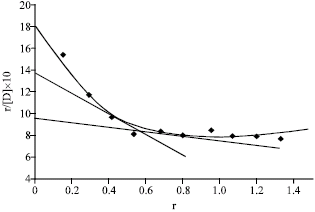
The curve thus obtained called scatchard plot. The scatchard plot when extrapolated on Y axis, gave an intercept nK, the intersection on X-axis representing n and the slope of line AB being k. Here, k is the affinity constant and n is the number of binding sites of protein binding.
Statistical analysis: The results were expressed as Mean± SEM values for each experiment. Differences in Mean values between experimental groups were analyzed by unpaired t-test. A probability value less than 0.05 (p<0.05) was defined to be significant.
RESULTS
From the Fig. 2, protein binding versus concentration of caffeine shows that at low concentration, the percentage of protein binding decreases with the increase in concentration of the drug. But at higher concentrations, the percentage attains a steady plateau indicating the saturation zone for the binding of caffeine to Bovine Serum Albumin (BSA). In the present study, the percentage of binding of caffeine to BSA at saturation level is about 93.
From the Fig. 3, it was found that the highest percentage of protein binding of caffeine at saturation level was about 85 in presence of gliclazide. By comparing this with that of caffeine alone, it can be inferred that gliclazide has significant effect on the protein binding of caffeine. This is obviously due to a good affinity of the complex and also gliclazide for the protein. The significant lowering of protein binding of caffeine due to gliclazide interference indicates that binding of gliclazide is also site specific.
From the Fig. 4, it was found that the highest percentage of protein binding of caffeine at saturation level was about 83 in presence of metformin HCl. So, by comparing this with that of caffeine alone, it can be inferred that metformin HCl has significant effect on the protein binding of caffeine (Amran and Hossain, 1998; Amran et al., 1999).
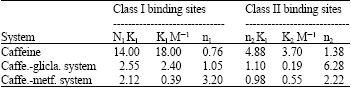
The scatchard plots show at least two classes of binding sites (class I and II, the warfarin and the diazepam sites, respectively). The number of binding sites n1 and n2 for class I and class II and affinity constants k1 and k2 for these classes have been calculated from scatchard plots. Numbers of binding sites were obtained by dividing the intercept (nk) by slope (k) of the straight lines. The values for affinity constants associated with respective class of binding sites were obtained directly from the slope of the straight lines (Scatchard, 1949).
From scatchard plots (Fig. 5), the number of binding sites for caffeine alone in BSA was found to be 0.76 and 1.38 for class I and II, respectively. The affinity constants k1 and k2 associated with class I and class II were 18 and 3.70, respectively.
From scatchard plots (Fig. 6), the number of binding sites for caffeine-gliclazide system in BSA was found to be 1.05 and 6.28 for class I and II, respectively. The affinity constants k1 and k2 associated with class I and II were 2.40 and 0.19, respectively.
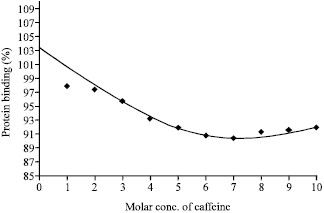 | |
| Fig. 2: | Protein binding of caffeine alone |
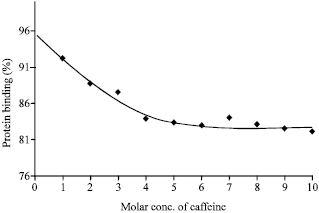 | |
| Fig. 3: | Protein binding of caffeine in presence of gliclazide (1:1 mixture) |
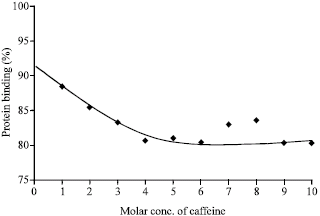 | |
| Fig. 4: | Protein binding of caffeine in presence of metformin HCI (1:1 mixture) |
From scatchard plots (Fig. 7), the number of binding sites for caffeine-metformin HCl system in BSA was found to be 3.20 and 2.22 for class I and II, respectively. The affinity constants k1 and k2 associated with class I and II were 0.39 and 0.55, respectively (Amran and Hossain, 1998; et al., 1999).
 | |
| Fig. 5: | Scatchard plot for protein binding of caffeine alone |
 | |
| Fig. 6: | Scatchard plot for protein binding of caffeine in presence of gliclazide (1:1 mixture) |
 | |
| Fig. 7: | Scatchard plot for protein binding of caffeine in presence of metformin HCl (1:1 mixture) |
According to Table 1, it was found that gliclazide causes a decrease in associated affinity constants but increases the number of binding sites in the mixed condition. Metformin HCl lowers the associated affinity constants for both class I and II binding sites. It increases the number of binding sites largely for class I binding sites and shortly for class II binding sites of caffeine.
| Table 1: | The values for number of binding sites and affinity constants |
 | |
DISCUSSION
Due to decrease in affinity to plasma protein binding, there is an increase in the apparent volume of distribution (Vd) of the drug because affinity of a drug for protein binding is a limiting factor of the distribution of the drug (Hansten and Horn, 1989). In other words due to increase in affinity, the Vd decreased. Vd can be calculated by dividing the amount of drug in the body by the plasma concentration. Since, the apparent volume of distribution increases in the both cases it is a matter of great concern that a concurrent application of caffeine and gliclazide and metformin HCl should be considered only after through in vivo studies.
The aim of the study was to infer about the combination therapy of oral antidiabetic gliclazide and metformin HCl with caffeine but present finding indicates that gliclazide and metformin HCl decrease the percentage of protein binding of caffeine i.e., increase the free plasma concentration of caffeine which may give toxic effects. Therefore, we infer that the combination therapy of caffeine with gliclazide and caffeine with metformin HCl may not be safe. Because such type of combination therapy may change the pharmacokinetic and pharmacodynamic properties of caffeine.
From this study, we can infer that the concurrent therapy of caffeine with either gliclazide or metformin HCl may increase hepatic first pass effect. Thus renal clearance of the drug and its therapy may alter the half life (t1/2) of the drug. Thus due to increase in concentration of free drug or decrease in affinity for protein, the pharmacological effects of drug will increase if the concentration of the drug remains within Minimum Effective Concentration (MEC) and Minimum Toxic Concentration (MTC), though t1/2 is shortened. But, if the concentration exceeds the MTC then toxicity appears (Tillement et al., 1974).
Salam and Hossain (2001), Milon and Hossain (2009) and Rahman and Hossain (2008) are engaged in the study of interaction between oral anti-diabetic drugs and other agents. In such studies, most of the agents used did not interact strongly with the oral anti-diabetic agents but in the present study, metformin HCl decreased the percentage of protein binding of caffeine. This will increase the free plasma concentration of the caffeine which may affect pharmacokinetic and pharmacodynamic activities of drug. Such a change in the pharmacokinetic and pharmacodynamic behavior might not be beneficial to the patients.
CONCLUSION
It was observed that gliclazide and metformin HCl lowered the affinity of protein binding of caffeine; hence an increase in volume of distribution of caffeine might be occurred. Therefore, it can be inferred that care and monitoring should be practiced during administration of caffeine-gliclazide and caffeine-metformin HCl complexes or concurrent administration of caffeine with gliclazide and metformin hydrochloride.
ACKNOWLEDGMENT
The authors are grateful to the authority of the Orion Laboratories Ltd., Dhaka, Bangladesh, for providing the studied drugs.
REFERENCES
- Amran, M.S., A.H.M.R. Bari and M.A. Hossain, 2006. In vitro and in vivo interactions of diltiazem with ibuprofen and naproxen in aqueous medium and rabbits. Dhaka Univ. J. Pharm. Sci., 5: 25-28.
Direct Link - Amran, M.H., M. Rahatuzzamn and M.A. Hossain, 2006. The synergism of diltiazem and nifedipine in their antihypertensive functions in the animal model and the case of coadministration of ketotifen fumerate and potassium nitrate with them. J. Biol. Sci., 14: 61-67.
Direct Link - Amran, M.S., S.N. Morshed, M.J.A. Khandakar, M.M. Rahman, M.M. Rahman and M.A. Hossain, 2008. The in vitro effects of atenolol and zinc chloride on the protein binding of amlodipine in aqueous medium. Dhaka Univ. J. Pharm. Sci., 7: 15-21.
Direct Link - Rashidul Bari, A.H.M., A.T.M. Zafrul Azam, M.S. Amran and M.A. Hossain, 2000. In vivo effects of ibuprofen and naproxen on the plasma concentration of diltiazem in rabbits. Pak. J. Biol. Sci., 3: 555-557.
CrossRefDirect Link - Kobayashi, K., M. Kimura, T. Sakoguchi, A. Hase, A. Matsuoka and S. Kaneko, 1984. Pharmacokinetics of gliclazide in healthy and diabetic subjects. J. Pham. Sci., 73: 1684-1687.
CrossRefPubMedDirect Link - Sweetman, S.C., 2005. Martindale, The Complete Drug Reference. 34th Edn., Pharmaceutical Press, Chicago, London, pp: 1544.
CrossRef - Sambol, N.C., J. Chiang, M. O'Conner, C.Y. Liu and E.T. Lin et al., 1996. Pharmacokinetics and pharmacodynamics of metformin in healthy subjects and patients with non-insulin-dependent-diabetes-mellitus. J. Clin. Pharmacol., 36: 1012-1021.
PubMed - Scatchard, G., 1949. The attractions of proteins for small molecules and ions. Ann. N. Y. Acad. Sci., 51: 660-672.
CrossRefDirect Link - Graham, G.G., J. Punt, M. Arora, R.O. Day and M.P. Doogue et al., 2011. Clinical pharmacokinetics of metformin. Clin. Pharmacokinet., 50: 81-98.
CrossRefDirect Link