
J.F. Gomez-Leyva
Laboratorio de Biologia Molecular, Instituto Tecnologico de Tlajomulco, Jalisco. Tlajomulco de Zuniga, Jalisco, Mexico
J. Lara-Reyna
Colegio de Postgraduados Campus Campeche, Campeche, Campeche Mexico
L.V. Hernandez-Cuevas
CICB, Universidad Autonoma de Tlaxcala, Tlaxcala, Tlaxcala, Mexico
J.P. Martinez-Soriano
CINVESTAV, Campus Guanajuato, Irapuato, Guanajuato, Mexico
Journal of Biological Sciences
Year: 2008 | Volume: 8 | Issue: 3 | Page No.: 563-569







ABSTRACT
Specific primers and Polymerase Chain Reaction (PCR) assays that identify Arbuscular Mycorrhizal (AM) fungi Glomus intraradices were developed. Monoxenic cultures of fungi G. intraradices and Gigaspora gigantea in association with Ri T-DNA transformed carrot roots were established in order to obtain fungal DNA free of host and others contaminants. RAPD analysis using 10 AM fungi from genera Glomus, Gigaspora and Acaulospora allowed the determination of two amplified fragments that were specific to G. intraradices. The DNA fragments were cloned, sequenced and subsequently used to design SCAR species-specific primers. A set of primers, GIN630F and GIN630R, drove the amplification of a 630 bp fragment specific for G. intraradices, which was absent when DNA of other AM fungi or plants were used as templates. The assay allowed the detection of G. intraradices in colonized roots of carrot. The SCAR-based protocol described here may be a tool of great value in studies of Glomeromycota`s molecular systematic and ecology.
PDF Abstract XML References Citation
How to cite this article
J.F. Gomez-Leyva, J. Lara-Reyna, L.V. Hernandez-Cuevas and J.P. Martinez-Soriano, 2008. Specific Polymerase Chain Reaction-Based Assay for the Identification of the Arbuscular Mycorrhizal Fungus Glomus intraradices. Journal of Biological Sciences, 8: 563-569.
DOI: 10.3923/jbs.2008.563.569
URL: https://scialert.net/abstract/?doi=jbs.2008.563.569
DOI: 10.3923/jbs.2008.563.569
URL: https://scialert.net/abstract/?doi=jbs.2008.563.569
INTRODUCTION
The Phylum Glomeromycota represents an interesting biological group because all its members fall in one of two types of symbiotic mutualistic associations. One of them is constituted for the monotypic family Geosiphonaceae whose only member Geosiphon pyriformis forms a peculiar symbiosis named endocytobiosis with cyanobacteria belonging to the genus Nostoc (Gehrig et al., 1996). The other symbiotic type is the very common arbuscular mycorrhiza, which is formed by the rest of the fungal species. Recent data establish that AM is associated to the roots of 80% of plant species, which indicates the complexity in origin, evolution and diversification of this group (Wang and Qui, 2006).
Traditionally, the taxonomic identification of AM fungi has been based on the morphological features of spores. Undoubtedly these structures contain important taxonomic information (Sieverding and Oehl, 2006) and the morphological approach to the identification of species although useful to estimate biodiversity it is also limited and controversial. The spores are persistence structures basically formed under unfavorable environmental conditions; their presence may be resultant of past events and not to show prevalent conditions during the sampled period (Lovelock and Ewel, 2005). Moreover, because most of the spores are produced outside the roots it could lead to an underestimation of the biodiversity of AM fungi on field samples. In order to solve these problems recent research has focused on the selection and implementation of tools to facilitate an accurate and reproducible identification of AM fungus (Reddy et al., 2005).
Molecular markers have been specifically developed for the detection and identification of pathogens with impressive accuracy. Furthermore, they have been successfully used in detecting fungi (Nazar et al., 1991; Simon et al., 1992; Berbee and Taylor, 1995). Nevertheless, the sequences used to design the assays have been targeted to ribosomal genes and consequently, they are universal and in some cases no species-specific (Schübler et al., 2001a), but genera or superior taxonomic ranks. Studies on Glomus intraradices have proven that the ITS region of the ribosomal genes is variable; therefore this fact compromises the specificity of the test (Jansa et al., 2002; Reddy et al., 2005).
An interesting strategy to develop species-specific molecular markers is based on the isolation, further sequencing of DNA fragments amplified by RAPD PCR and the use of designed primers to specific target sequences that may be unique for a species. These molecular markers are known as Sequence Characterized Amplified Region or SCARs (Paran and Michelmore, 1993). This approach has been applied to the identification of different species of fungi, including some ectomycorrhizal (Gandeboeuf et al., 1997). In contrast, this is not an easy task for MA fungi due to the mutualistic symbiosis with the plant, which makes difficult the isolation of uncontaminated DNA. The use of in vitro root-organ cultures (Bécard and Piché, 1992) may overcome this obstacle; however, there have been very few species on which this system has been successful obtained. The objective of this research was to establish monoxenic cultures of the fungi G. intraradices and Gigaspora gigantea to obtain pure DNA to further develop a specific test designed to identify the species by the use of a SCAR marker. This method would help to determinate accurately the identity of these species in natural ecosystems; moreover, it may contribute to the estimation of the relative abundance of the fungus into the roots.
MATERIALS AND METHODS
Fungal material and strains: The AM fungi Glomus intraradices Schenck and Smith strain 0046TLX03, G. intraradices strain BEG 144, G. caledonium (Nicol. and Gerd.) Trappe and Gerd. strain BEG 20, G. claroideum Schenck and Smith strain 0003TLX01, G. etunicatum Becker and Gerdemann strain 0004MOR01, G. fasciculatum (Thaxter) Gerd. and Trappe emend. Walker and Koske, G. mosseae (Nicol. and Gerd.), Gigaspora gigantea (Nicol. and Gerd.) Gerd and Trappe strain 0033TLX06, G. margarita Becker and Hall strain 0036TLX06, Scutellospora dipurpurasens Morton and Koske strain 0020TLX06, S. pellucida (Nicol. and Schenck) Walker and Sanders strain 0018TLX06, Acaulospora lacunosa Morton strain BEG 78, A. laevis Gerdemann and Trappe strain BEG 13, A. longula Spain and Schenck strain BEG 8 and A. spinosa Walker and Trappe strain 0039TLX01 were obtained in pure pot culture from AM fungi collections of the CICB from the Universidad Autónoma de Tlaxcala (TLX and MOR codes) and the European Bank of Glomales (BEG codes) and kept in soil at 4 °C.
Production of hairy roots and monoxenic cultures: The strains of Agrobacterium rhizogenes LBA 9402 and agropine type AR12 were used for DNA transformation of carrot (Daucus carota L.) to which the binary vector pBI121 was inserted. Both strains harbored the wild type Ri plasmid. Bacterial cells were grown at 28 °C in YEB (10 g L-1 yeast extract, 5 g L-1 beef extract, 5 g L-1 peptone, 5 g L-1 saccharose, 0.49 g L-1 MgSO4.7H2O supplemented with 50 mg L-1 of riphampycin plus 100 mg L-1 of kanamycin) to an OD 600 of 0.4. The infection process was performed on transversal disks of carrot co-cultivated with 0.5 mL of the bacterial suspension (OD 600 = 1) for 24 h in the darkness. Tissues were transferred to medium MS (Murashige and Skoog, 1962) supplemented with 500 mg L-1 of cephotaxyme and incubated at 25 °C. The bacteria-free hairy roots were removed and transferred to minimum media (M) (Bécard and Piché, 1992) and maintained as clones. Spores of G. intraradices and G. gigantea were extracted by wet sieving and surface sterilized by treatment with 2% Chloramine T plus 0.1% Tween 20 for 15 min at 4 °C, vacuum was applied and the procedure was repeated twice. The spores were transferred to an antibiotic solution (100 mg L-1 of gentamycin sulphate, 2000 mg L-1 streptomycin sulphate) for 24 h at 4 °C. After this treatment, the spores were rinsed with double distilled water and gently distributed over a plate containing M medium and incubated at 25 °C for 3-4 days. Germinated spores were then transferred to the proximity of the transformed Ri T-DNA carrot roots having active growth to stimulate the colonization. In order to obtain a massive production of spores, the dual system developed by St-Arnaud et al. (1996) was used.
PCR analysis of transformed roots: Genomic DNA of the different root clones was extracted according to Doyle and Doyle (1990). Twenty nanogram of genomic DNA was used in a reaction volume of 25 μL containing 15 mM Tris-HCl (pH 8), 0.1% Triton X-100, 50 mM KCl, 1.5 mM MgCl2, 100 μM of each dNTP, 2.5 U de Taq DNA polymerase (Promega, Madison, WI) and 10 pmol of each of the primers ROLB1 (5´ATG GAT CCC AAA TTG CTA TTC CTT CCA CGA), ROLB2 (5´ TTA GGC TTC TTT CTT CAG GTT TAC TGC AGC), VIRD1 (5´ATG TCG CAA GGA CGT AAG CCCA) and VIRD2 (5´GGA GTC TTT CAG CAT GGA GCA A) (Hamill et al., 1991). These primers drove the amplification of 780 bp and 450 bp fragments from the genes rol B and vir D1 of Agrobacterium, respectively. The amplification was performed using an MJ Research PT-100 thermocycler (MJ Research, Watertown, MA) using the following profile: an initial denaturation step of 94 °C/3 min, followed by a 25 cycles of 94 °C/1 min, 55 °C/1 min, 72 °C/1.5 min and a final extension step of 72 °C/7 min then held at 4 °C. The amplified products were fractionated in 1.2% agarose gels.
Fungal DNA extraction and RAPD analysis: Spores of G. intraradices and G. gigantea were obtained from the distal part of the monoxenic culture by dissolving Phytagel in 10 mM sodium citrate and crushed in 40 μL of TE buffer (10 mM Tris-HCl pH 8.1, 1 mM EDTA) and heated to 95 °C for 20 min in 40 μL of 30% w/v Chelex-100 resin (BioRad). Genomic DNA was separated from cellular debris by centrifugation at 14,000 rpm for 1 min; the resulting extract was diluted and used immediately for use in the PCR assays. Spores of the other AM fungi species were isolated from the soil of propagation pots using the wet sieving and decantation process (Gerdemann and Nicolson, 1963) and were later surface sterilized by treatment with 2% Chloramine T and antibiotics solutions. DNA extraction of these fungal species was conducted according to Lee and Taylor (1991). RAPD reactions were performed in a total volume of 50 μL containing 10 mM Tris-HCl (pH 8.8), 50 mM KCl, 1.5 mM MgCl2, 0.1% Triton X-100, 100 μm each of dNTP (Invitrogen, Carslbad, CA), 0.5 μM each random 10-mer (Bio-Synthesis Co., Lewisville, TX), 20 ng of genomic DNA and 2 units of Taq DNA polymerase (Promega, Madison, WI). Thermal Cycler MJ Research PTC-100 was programmed for an initial denaturation at 94 °C/3 min, followed by 35 cycles of 94 °C/1 min, 36 °C/1 min, 72 °C/2 min and a final extension at 72 °C/7 min. Amplification products were separated in 1.4% agarose gels and stained with ethidium bromide; the DNA bands were visualized under UV light and photographed. Every experiment was performed by duplicate.
Cloning and sequencing of RAPD markers: Polymorphic RAPD fragments amplified from G. intraradices and G. gigantea were purified using the Wizard PCR Preps kit (Promega, Madison, WI) and cloned into the pGEM-T Easy vector (Promega, Madison, WI) as recommended by the manufacturer. The recombinant vectors were used to transform competent Escherichia coli cells DH5α. The selection of recombinants was performed by PCR using white colonies directly as source of template DNA that was amplified utilizing the RAPD primers employed for the first amplification. Plasmid DNA from recombinant colonies was purified using the High Pure Plasmid Isolation kit (Boehringer Ingelheim, GmbH, Germany). Insert size was verified by EcoR I digestions, followed by 1.5% agarose gel fractionation. The complete sequence of each cloned fragment was obtained by the use of an automated sequencing robot ABI PRISM 377 (Applied Biosystems, Foster City, CA). DNA sequences were compared by alignment by the DNASIS V 2.0 program for the Macintosh system. DNA sequences for G. intraradices were deposited to the GenBank databases.
Design of SCAR primers and PCR conditions: The search for DNA similarities was performed using the BLAST and BLASTX programs from the NCBI network service (Altschul et al., 1997). For each RAPD fragment several SCAR primers were designed using the complete DNA sequence for the non-occurrence of secondary structures. The absence of cross hybridization was checked using the PrimerSelect 3.11 software For Windows (DNAStar, Lasergene, Madison, WI). Primers were designed with GC content of 50-60% and synthesized by Invitrogen. The PCR specific assays were carried out in a total volume of 25 μL containing approximately 5 ng DNA, 1X reaction buffer (Promega), 200 μM of each dNTP (Invitrogen), 1.5 mM MgCl2, 20 pmol SCAR primers, 2.5 U Taq polymerase (Promega). Amplifications with SCAR primers were performed in the MJ-Research PTC-100 as follows: initial denaturation at 94 °C/2 min, followed by 30 cycles at 94 °C/1 min, 53-58 °C/1 min, 72 °C/1 min and then a final extension at 72 °C/7 min. As proper controls, DNA of every species was amplified by using the universal ITS primers ITS1 and ITS4 as described by White et al. (1990). Amplification products were fractionated by gel electrophoresis in 1.4% agarose gels in 1X TAE buffer (40 mM Tris-acetate, 1 mM EDTA, pH 8), stained with ethidium bromide and visualized under UV light.
RESULTS AND DISCUSSION
Carrot roots transformation and monoxenic culture: Spores are the only differentiated biologically structures of AM fungi that could be studied outside the host. Even though spores need to be disinfected to remove contaminants, the presence of multiple nuclei makes the interpretation of the RAPD analysis complex. In order to avoid that, we established monoxenic cultures of G. intraradices and G. gigantea. The culture system requires the maintenance of roots in an autonomous and undefined stage of continuous growth under controlled conditions. In this research, we used two types of A. rhizogenes strains such as LBA9402 and AR12 for the production of transformed roots (Fig. 1A). Both strains transfer their T-DNA and induced hairy roots free of bacterial cells. Consequently, roots obtained with strain LBA9402 agropine-manopine type resulted in better vigor and branching growth compared with roots produced with
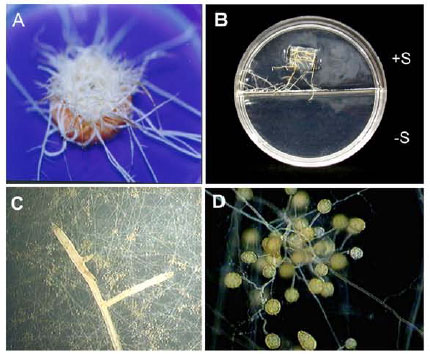 |
| Fig. 1: | Monoxenic culture of Glomus intraradices with transformed roots. (A) Ri T-DNA transformed roots growth on carrot disc. (B) Dual culture of carrot root and fungus in a two compartment Petri dish: –S, M medium minus saccharose; +S, plus saccharose. (C) Massive production of spores by G. intraradices grown in M medium and (D) Details of vegetative spores of G. intraradices |
strain AR12. This may be due to a higher accumulation of auxins in roots (Nin et al., 1997). On the other hand, the disinfection method of spores reduced contamination to a 10% with a germination percentage of 70% of both species in agar-water. There are reports about several factors influence the in vitro germination rates of the spores, such as radical exudates, flavonoids, pH conditions, presence of CO2, low temperature storage and physiological status of the spore (Bécard et al., 1992; Chabot et al., 1992; Poulin et al., 1993; Juge et al., 2002). In our study germination was successful probably due to the optimal physiological situation of the spores or the disinfection process itself without need to add external components. Four weeks later of the in vitro infection initiation, a massive development of mycelium was observed on the medium accompanied by formation of Branched Absorbing Structures (BAS) as described by Bago et al. (1998).
The spore formation under the dual system (Fig. 1B) as described by St-Arnaud et al. (1996) started four months posterior to the inoculation of G. intraradices with an average of 800 spores per dish (Fig. 1C, D), whereas G. gigantea only produced 40 spores per dish during the same period.
RAPD analysis and PCR detection: Only three of the twenty five primers produced consistent amplification patterns. Primer 5´-TGCAGCACCG (Bio-synthesis 70-09), which has a 70% content of G+C, drove the amplification of two specific fragments of 950 and 650 bp for G. intraradices (Fig. 2). These fragments were cloned and sequenced (GenBank accession numbers AY244447 and AY244448). The terminal ends of the sequenced fragments perfectly matched the sequence of the 10-mer used for the PCR amplification. From all stock of AM fungi species used in this study, only G. claroideum, G. fasciculatum, G. gigantea and G. margarita could be amplified, all other produce no satisfactory results, which reinforce that AM fungi DNA extraction and amplification is a difficult task for almost all species, specially if these species are not been propagated monoxenically.
Based on the disclosed DNA sequences, several set primers were designed, synthesized and used to optimize PCR assay and the amplification of a unique fragment to accurately identify G. intraradices. Primers GIN930F (5` TGC AGC ACC GCC TCC ACC) and GIN930R (5` TGC AGC ACC GTC GCT TGT TA) drove the amplification of an expected 930 bp fragment. This method encouraged the detection of G. intraradices strain 0046TLX03 in fragments of infected in vitro roots of D. carota and in vivo roots of Sorghum sp. colonized with G. intraradices strain BEG144 (data not shown).
In addition, consistent results were obtained with primer set GIN630F (5` GCA CCG CAA GTT AAG TAC
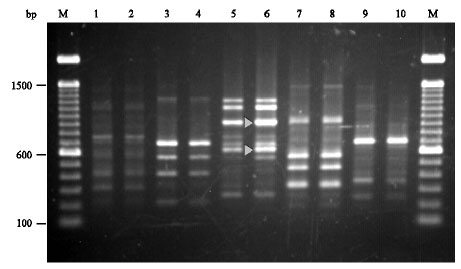 |
| Fig. 2: | Random amplified polymorphic DNA (RAPD) fingerprints using genomic DNA of mycorrhizal arbuscular fungi. Lanes 1-2 Glomus claroideum, 3-4 G. fasciculatum, 5-6 G. intraradices 0046TLX03, 7-8 Gigaspora gigantea, 9-10 G. margarita. M 100 bp DNA ladder. Arrows indicate specie-specific and reproducible RAPD bands of 950 and 650 bp that were converted into SCAR markers |
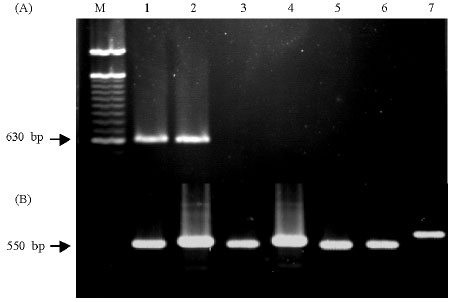 |
| Fig. 3: | (A) Agarose gel electrophoresis of PCR products obtained using the Glomus intraradices-specific primers GIN630F/R designed in this study. (B) Agarose gel electrophoresis of ITS 1/4 primers used as internal controls. M 100 bp DNA ladder. Lanes (1) genomic DNA from Glomus intraradices 0046TLX03, (2) carrot root colonized with G. intraradices BEG144, (3) G. fasciculatum, (4) G. claroideum, (5) G. mosseae, (6) Acaulospora laevis, (7) Daucus carota |
CCA AC) and GIN630R (5` CCG TGA TCA TGA TGT CTC AGG TT). Annealing temperatures of 54 °C were used to produce the expected fragment of 630 bp. Lower temperatures allowed the amplification of an unspecific fragment of 800 bp. The test proved to be highly specific to G. intraradices and no amplified DNA was observed when genomic DNA from others AM fungi was used as template (Fig. 3A). Controls DNA of every species were successfully amplified by using the universal ITS primers (Fig. 3B).
There are reports of PCR-based tests that have been presented as specific for several fungi of the order Glomerales (cited as Glomales by Simon, 1996). Many of them have been developed by using ribosomal DNA as template such as the VANS1 primer set proposed by Simon et al. (1993). Recently, it has been demonstrated that these primers are not specific and that primer homologous sequences are absent in at least 88 of the MA fungi analyzed (Lloyd-MacGilp et al., 1996; Schübler et al., 2001b; Sanders, 2003). Moreover, for G. mosseae alone, at least 23 sequences with homology varying from 66 to 98% have been reported by Antoniolli et al. (2000).
The nucleotide sequences corresponding to SCAR fragments reported here may be of vital importance for the development of quantitative PCR assays for the study of G. intraradices. The distribution of the AM fungus in the host roots could be now elucidated and consequently a better understanding of spore abundance in the soil and infection process.
This research has successfully demonstrated the use of monoxenic cultivation of two AM fungi and its further use to analyze RAPD patterns in order to develop specific PCR tests based on the selection of unique SCAR sequences for different species. Furthermore, the use of this approach contributed to the accurate detection and identification of G. intraradices on in vitro and in vivo tests. The test offers promising use for further studies on the ecology of the mycorrhiza-plant interaction in nature.
ACKNOWLEDGMENTS
This study was supported by funds from Consejo Nacional de Ciencia y Tecnología (CONACyT) grant number 26357 B. We thank Juan Carlos Ochoa-Sánchez, Magali Hernandez-Valencia and Juan Enrique Cortés-Valle for his technical support.
REFERENCES
- Altschul, S.F., T.L. Madden, A.A. Schaffer, J. Zhang, Z. Zhang, W. Miller and D.J. Lipman, 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucl. Acids Res., 25: 3389-3402.
CrossRefPubMedDirect Link - Antoniolli, Z.I., D.P. Schachtman, K. Ophel-Keller and S.E. Smith, 2000. Variation in rDNA ITS sequences in Glomus mosseae and Gigaspora margarita spores from a permanent pasture. Mycol. Res., 104: 708-715.
Direct Link - Bago, B., C. Azcon-Aguilar, A. Goulet and Y. Piche, 1998. Branched absorbing structures (BAS): A feature of the extraradical mycelium of symbiotic arbuscular mycorrhizal fungi. New Phytol., 139: 375-388.
CrossRef - Becard, G., P. Douds and P. Pfeffer, 1992. Extensive in vitro hyphal growth of vesicular-arbuscular mycorrhizal fungi in the presence of CO2 and flavonols. Applied Environ. Microbiol., 58: 821-825.
Direct Link - Berbee, M.L. and J.W. Taylor, 1995. From 18S ribosomal sequence data to evolution of morphology among the fungi. Can. J. Bot., 73: S677-S683.
CrossRef - Chabot, S., R. Bel-Rhlid, R. Chenevert and Y. Piché, 1992. Hyphal growth promotion in vitro of the VA mycorrhizal fungus, Gigaspora margarita Becker and Hall, by activity of structurally specific flavonoid compounds under CO2-enriched conditions. New Phytol., 122: 461-467.
CrossRef - Gandeboeuf, D., C. Dupre, P.N. Roeckel-Drevet and G. Chevalier, 1997. Typing tuber ectomycorrhizae by polymerase chain reaction of the internal transcribed spacer of rDNA and the sequence characterized amplified region markers. Can. J. Microbiol., 43: 723-728.
PubMed - Gehrig, H., A. Schübler and M. Kluge, 1996. Geosiphon pyriforme, a fungus forming endocytobiosis with Nostoc (cyanobacteria), is an ancestral member of the Glomales: Evidence by SSU rRNA analysis. J. Mol. Evolu., 43: 71-81.
CrossRefDirect Link - Gerdemann, J.W. and T.H. Nicolson, 1963. Spores of mycorrhizal Endogone species extracted from soil by wet sieving and decanting. Trans. Br. Mycol. Soc., 46: 235-244.
CrossRefDirect Link - Hamill, D.J., S. Rounsley, A. Spencer, G. Todd and M.J. Rhodes, 1991. The use of polymerase chain reaction in plant transformation studies. Plant Cell Rep., 10: 221-224.
CrossRef - Jansa, J., A. Mozafar, T. Anken, R. Ruh, I. Sanders and E. Frossard, 2002. Diversity and structure of AMF communities as affected by tillage in a temperate soil. Mycorrhiza, 12: 225-234.
CrossRefDirect Link - Juge, C., J. Samson, C. Bastien, H. Vierheilig, A. Coughlan and Y. Piché, 2002. Breaking dormancy is spores of the arbuscular mycorrhizal fungus Glomus intraradices: A critical cold-storage period. Mycorrhiza, 12: 37-42.
Direct Link - Lloyd-MacGilp, S.A., S.M. Chambers, J.C. Dodd, A.H. Fitter, C. Walker and J.P.W. Young, 1996. Diversity of ribosomal internal transcribed spacers within and among isolates of Glomus mosseae related mycorrhizal fungi. New Phytol., 133: 103-111.
CrossRef - Lovelock, C.E. and J.J. Ewel, 2005. Links between tree species, symbiotic fungal diversity and ecosystem functioning in simplified tropical ecosystems. New Phytol., 167: 219-228.
Direct Link - Murashige, T. and F. Skoog, 1962. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol. Plant., 15: 473-497.
CrossRefDirect Link - Nazar, R.N., X. Hu, J. Schmidt, D. Culham and J. Robb, 1991. Potential use of PCR-amplified ribosomal intergenic sequences in detection and differentiation of Verticillium wilt pathogens. Physiol. Mol. Plant Pathol., 39: 1-11.
CrossRef - Nin, S., A. Bennici, G. Roselli, D. Mariotti, S. Schiff and R. Magherini, 1997. Agrobacterium-mediated transformation of Artemisia absinthium L. (wormwood) and production of secondary metabolites. Plant Cell Rep., 16: 725-730.
CrossRef - Paran, I. and R.W. Michelmore, 1993. Development of reliable PCR-based markers linked to downy mildew resistance genes in lettuce. Theor. Applied Genet., 85: 985-993.
CrossRef - Poulin, M., R. Bel-Rhlid, Y. Piche and R. Chenevert, 1993. Flavonoid release by carrot Daucus carota seedlings stimulate hyphal development of vesicular arbuscular mycorrhizal fungi in the presence of the optimal CO2 enrichment. J. Chem. Ecol., 19: 2317-2327.
CrossRef - Reddy, S.R., P.K. Pindi and S.M. Reddy, 2005. Molecular methods for research on arbuscular mycorrhizal fungi in India: Problems and prospects. Curr. Sci., 89: 1699-1709.
Direct Link - Sanders, I.R., 2003. Preference, specificity and cheating in the arbuscular mycorrhizal symbiosis. Trends Plant Sci., 8: 143-145.
Direct Link - Schüβler, A., D. Schwarzott and C. Walker, 2001. A new fungal phylum, the Glomeromycota: Phylogeny and evolution. Mycol. Res., 105: 1413-1421.
CrossRefDirect Link - Schuβler, A., H. Gehrig, D. Schwarzott and C. Walker, 2001. Analysis of partial Glomales SSU rRNA gene sequences: Implications for primer design and phylogeny. Mycol. Res., 105: 5-15.
Direct Link - Sieverding, E. and F. Oehl, 2006. Revision of Entrophospora and description of Kuklospora and Intraspora, two new genera in the arbuscular mycorrhizal Glomeromycetes. J. Applied Bot. Food Qual., 80: 69-81.
Direct Link - Simon, L., R.C. Levesque and M. Lalonde, 1993. Identification of endomycorrhizal fungi colonizing roots by fluorescent single-strand conformation polymorphism polymerase chain reaction. Applied Environ. Microbiol., 59: 4211-4215.
Direct Link - Simon, L., 1996. Phylogeny of the glomales: Deciphering the past to understand the present. New Phytol., 133: 95-101.
CrossRefDirect Link - St-Arnaud, M., C. Hamel, B. Vimard, M. Caron and J.A. Fortin, 1996. Enhanced hyphal growth and spore production of the arbuscular mycorrhizal fungus Glomus intraradices in an in vitro system in the absence of the host roots. Mycol. Res., 100: 328-332.
CrossRef - Wang, B. and Y.L. Qiu, 2006. Phylogenetic distribution and evolution of mycorrhizas in land plants. Mycorrhiza, 16: 299-363.
CrossRefPubMedDirect Link - White, T.J., T.D. Bruns, S.B. Lee and J.W. Taylor, 1990. Amplification and Direct Sequencing of Fungal Ribosomal RNA Genes for Phylogenetics. In: PCR Protocols: A Guide to Methods and Applications, Innis, M.A., D.H. Gelfand, J.J. Sninsky and T.J. White (Eds.), Academic Press, San Diego, CA, USA, ISBN-13: 9780123721808, pp: 315-322.
CrossRefDirect Link