
M. Tehranipour
Department of Biology, Faculty of Science, Islamic Azad University, Mashhad Branch, Mashhad, Iran
M.R. Khakzad
Medical University, Islamic Azad University, Mashhad Branch, Mashhad, Iran
Journal of Biological Sciences
Year: 2008 | Volume: 8 | Issue: 6 | Page No.: 1027-1032







ABSTRACT
The present study has been undertaken the effects of maternal diabetes on Hippocampus structure 1 day neonate individual`s rats from diabetic mothers in both control and diabetic groups. Diabetes was induced by stereptozotocin (60 mg kg-1) given by a single intraperitoneal injection to female Wistar rats. Control rats were injected with phosphate buffered saline. In neonates brains rapidly were removed and in all sample the number of neurons in CA1, CA2, CA3 was measured via stereological method in both control and experimental groups. Statistical analysis determines that there is a meaningful reduction in number of neurons in CA3 in neonate of diabetic mothers (p<0.05). That may be the reason for memory problem in these neonates.
PDF Abstract XML References Citation
How to cite this article
M. Tehranipour and M.R. Khakzad, 2008. Effect of Maternal Diabetes on Hippocampus Neuronal Density in Neonatal Rats. Journal of Biological Sciences, 8: 1027-1032.
DOI: 10.3923/jbs.2008.1027.1032
URL: https://scialert.net/abstract/?doi=jbs.2008.1027.1032
DOI: 10.3923/jbs.2008.1027.1032
URL: https://scialert.net/abstract/?doi=jbs.2008.1027.1032
INTRODUCTION
Pathological changes in the central nervous system of diabetic animals and human are usually subtle in the brain (Polanco et al., 2005) selective vulnerability was reported especially in the cerebral cortex and in the hypothalamus (the preoptic-suprachiasmatic nuclei) in diabetic rats (Luo et al., 2002).
The hippocampus is a important structure for memory processing. It is a particularly vulnerable and sensitive region of the brain that is also very important for declarative and spatial learning and memory. Hippocampal neurons are vulnerable to seizures, strokes and head trauma, as well as responding to stressful experiences. At the same time they show remarkable plasticity, involving long-term synaptic potentiation and depression, dendrite remodeling (synaptic turnover and neurogenesis in the case of the dentate gyrus (McEwen, 1999). The hippocampus has been implicated in certain short-term memory. Indeed hippocampal lesions often produce short-term memory deficits (Mumby et al., 2002). The hippocampus is preferentially susceptible to a wide variety of toxic insults and disease processes, including hypoxia-ischemia and hypoglycemia (Piotrowski et al., 2001). Metabolic diseases such as diabetes and obesity have been associated with increased vulnerability to stress (Damasceno et al., 2002) and cognitive dysfunction (Nazer and Ramírez, 2000). Diabetes mellitus can lead to functional and structural deficits in both the peripheral and central nervous system. The pathogenesis of these deficits is multifactor and may involve microvascular dysfunction and oxidative stress (Patil et al., 2006). Cognitive deficits are also reported to occur in animal models of diabetes (Stroptozotocin induced)which can be prevented, but not fully reversed by insulin treatment (Kuhad et al., 2008). Diabetes also induced morphological changes in the presynaptic mossy fiber terminals (MFT) that form excitatory synaptic contacts with the proximal CA3 apical dendrites (Magariños and McEwen, 2000).
Oxidative stress induced by chronic hyperglycemia contributes to cerebrovascular complication in diabetes (Damasceno et al., 2002). Also diabetes mellitus is associated with an increased risk for cerebrovascular disease (Aragno et al., 2002). Accumulating data support the conclusion that oxidative stress induced by chronic hyperglycemia plays a key role in both microvascular and macrovascular complications of diabetes, including stroke (Artola, 2008). Many deleterious events contribute to oxidative damage to neurons in diabetes: because of high levels of polyunsaturated lipids in the brain, direct lipperoxidation frequently occurs causing lipid membrane disruption and consequent neurodegeneration (Ristow, 2004).
Moreover, oxidative stress increase tissue levels of highly reactive and toxic substances and effects signal transduction pathways involved in neuronal and endothelial cell function. Primary diabetic encephalopathy is recognized as a late complication of both type 1 and type 2 diabetes (Kinney et al., 2003). Impairments in learning, memory, problem salving and mental and motor speed are more common in type 1 diabetic patients than in the general population (Deregnier et al., 2000). A diabetic duration dependent decline in cognitive function occurs independently of hypoglycemic episodes (DeBoer et al., 2005) and impaired intellectual and cognitive developments in type 1 diabetic children correlate with diagnosis at young age, male sex and metabolic status. Cognitive deficits (Ryan and Geckle, 2000) and poor performances in abstract reasoning and complex psychomotor functioning occur in type 2 diabetes. Learning and memory dysfunctions are more prominent in elderly type 2 diabetic patients (Sinclair et al., 2000). It has not been determined whether this is because of potentiation of the normal aging process, a function of diabetes duration, or both. Notably Alzheimer disease is twice as prevalent in the diabetic population as in nondiabetic subjects (Arvanitakis et al., 2005). Several recent studies have implicated abnormal function of the insulin/IGF axis in the early pathogenesis of Alzheimer`s disease. Insulin and IGF-1 are believed to regulate β-amyloid levels and tau phosphorylation (Gasparini and Xu, 2003).
Impaired spatial learning and memory occur in animal models of both type 1 and type 2 diabetes. In the hippocampus of STZ-induced rats, long-term potentiation is impaired, whereas long-term depression is enhanced indicating altered hippocampal synaptic plasticity, which are corrected by insulin treatment (Gispen and Biessels, 2000).
Pregnant women who suffer from diabetes are more likely to have a child with memory problems; according to a new study. Fetal brain iron deficiency occurs in human pregnancies complicated by diabetes mellitus or more common in the offspring of these pregnancies. The aim of present research was to induce maternal diabetes mellitus and to assess the effects of that on Hippocampus structure (CA1, CA2, CA3) and measuring the neuronal density.
MATERIALS AND METHODS
Female Wistar rats, weighting 200-250 g, were hosed under standard laboratory conditions and kept under natural 12 h light: 12 h dark cycle. The animals procured from Razi Animal House, were housed 2 per cage with free access to standard food and water. Rats were acclimatized to laboratory conditions before testing. Animal were divided into 2 different groups for estiminat the effect of maternal hyperglycemia in neonates.
| Group 1: | Normal-rats were not subjected to any procedures |
| Group 2: | Diabetes-under STZ injection |
All the experimental protocols were conducted in Faculty of Science, Islamic Azad University of Mashhad, Iran (2008). All chemical used in this study were purchased from Sigma (UK).
Induction of diabetes: Diabetes was induced in rats by a single injection of STZ (60 mg kg-1) freshly dissolved in citrate buffer (pH 4.5). Age-matched control animals were injected with citrate buffer. Diabetes was confirmed after 8 weeks; only the animals with blood glucose level above 400 mg dL-1 were included in the study.
The body weight was measured at the beginning and the end of the experiment. All animals were checked for glucose blood concentration at the beginning of the experiment. After 2 weeks from STZ-injection, as well as on the day before the of experiment.
Tissue collection: Animals were anesthetized with sodium pentobarbital (64 mg kg-1) and decapitated. The whole brain was removed and fixed in 10% paraformaldehyde. NaCl was added to the fixative to make the tissue float in order to overcome deformities during the fixation period. Paraffin embedded tissue blocks were sectioned at 7 mμ thickness coronally and stained with haematoxylin-eosin.
Measurement of neuronal density in hippocampus: Hemotoxylin-eosin-stained serial paraffin sections were prepared from 8 hippocampi from individual animals in each group. Regions of hippocampus (CA1 CA3) were identified according to paxinos and watson. Tissue blocks containing samples (brains) were serially cut throughout. Form several hundred sections per block, of each 20 section 3 serial sections were obtained. For example for the first series: 24, 25, 26th section and for the second series: 46, 47, 48th section and so on. Therefore we mounted every 3 section on a slide. At a practical level, stereological methods are precise tools for obtaining quantitative information about three-dimensional structures based mainly on observations made on sections (Gunderson et al., 1988). All experiments were performed a minimum of two times. Student`s t-test was used for comparison when only 2 groups were analyzed. Statistical significance was chosen as p<0.05. All results are reported as Mean ± SEM.
RESULTS AND DISCUSSION
Two weeks after STZ-injection the rat`s blood glucose level was higher than 400 mg dL-1. At the end of the 10th week, the diabetic rats showed an increase in blood glucose level (470 ± 18 mg dL-1).
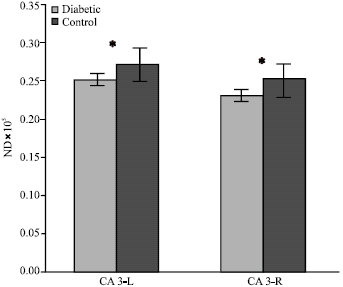 | |
| Fig. 1: | Neuronal density CA3 in neonate from diabetic mothers compare to control The values are presented as Means ± SEM n = 8 *p<0.05. Student`s t-test compare pups from Diabetic dams with pups from controls. CA3-L: CA3 in left hemisphere. CA3-R: CA3 in right hemisphere |
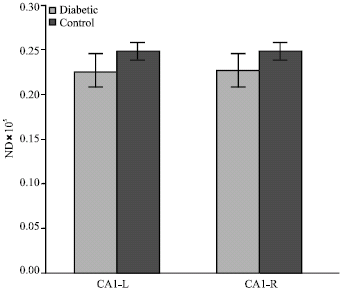 | |
| Fig. 2: | Neuronal density CA1 in neonate from diabetic mothers compare to control. The values are presented as means ± SEM. n = 8. *p<0.05 Student`s t-test compare pups from Diabetic dams with pups from controls. CA1-L: CA1 in left hemisphere. CA1-R: CA1 in Right hemisphere |
The body weight of diabetic animals increased to (230-260 g) during the experimental time, whole in the normal to (270-340 g). In the urine, all diabetic rats demonstrated abnormal results: glucosuria (+++), ketone bodies (trace) and protein (trace). In the plasma all diabetic rats had remarkable increase in Creatinine, Uric acid, Urea, Triglycerides, Cholesterol. [P3+], [Ca2+] (p<0.05).
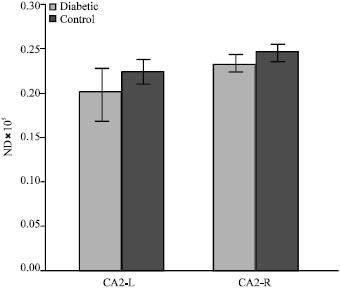 | |
| Fig. 3: | Neuronal density CA2 in neonate from diabetic mothers compare to control. The values are presented as means ± SEM. n = 8. *p<0.05 Student`s t-test compare pups from Diabetic dams with pups from controls. CA2-L: CA2 in left hemisphere. CA2-R: CA2 in Right hemisphere. |
Maternal hyperglycemia produced evoked significant neuronal loss in CA3 of neonate brains (Fig. 1). This decrease was 0.25 ± 0.01 in control to 0.23 ± 0.00 in neonates from diabetic mothers (p<0.05).
We estiminat left and right regions of hippocampus and the results were the same. Although there was remarkable decreased in CA1 and CA2 but they were not meaningful (Fig. 2, 3).
In CA1 the mean of neuronal density was 0.24 ± 0.01 in control to 0.22 ± 0.02 in neonates from diabetic mothers (p = 0.05).
ND in CA2 was 0.24 ± 0.00 in control to 0.23 ± 0.00 in experimental groups (p<0.08).
In morphometric study, there was more changes in neuronal shape in hippocampus regions of neonates from diabetic mothers camper to controls (Fig. 4).
In our morphometric studies on pyramidal cells in CA1, CA2 and CA3, it has been shown that the experimental maternal hyperglycemia in rats results neuronal loss and damage expressed maximally in CA3. Although in experimental groups there is a remarkable neuronal density decrease in all hippocampus sectors (Fig. 1-3), but it is meaningful in CA3 (Fig. 1).
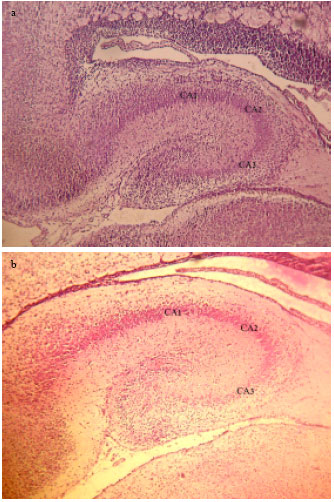 | |
| Fig. 4: | Photomicrograph of the brain section of neonate at the region of the midhippocampus. (a) hippocampus of neonate from diabetic mother and (b) Control. (X40) |
Diabetes causes morphometric neuronal changes (Sinclair et al., 2000). This observation has allowed us to postulate that the neuronal death in the infant from diabetic mothers proceeds on an apoptotic pathway (Sima and Li, 2005). Gestational conditions increase fetal iron demand for erythropoiesis beyond placental iron transport capacity. Diabetes mellitus can result in a 30-40% reduction in neonatal brain iron (Damasceno et al., 2002). Iron in the form of cytochoromes is a required component of cellular oxidative metabolism in the brain and is thus essential for normal neuronal function (Ornoy, 2007).
A higher prevalence of cognitive impairment has been documented in infants born to mothers with hyperglycemia gestational conditions (Gao and Gao, 2007). Iron deficiency could have a direct effect on brain development or could potentiate the effects of other adverse prenatal events. The cognitive nature of the long- term impairments of infants of diabetic mothers (Nold and Georgieff, 2004) and growth-retarded infants suggests that factors targeting the developing hippocampus may be particularly important. The hippocampus in necessary for normal cognitive function, especially for processing recognition memory and transferring short-term memory items into long-term storage (McEwen, 1999).
In humans and animal models, the hippocampus appears to be particularly vulnerable to prenatal hypoxia-ischemia an event that occurs more commonly in gestations complicated by maternal diabetes mellitus or fetal growth retardation. We hypothesized that prenatal brain iron deficiency that occur in diabetic pregnancy increases the vulnerability of the hippocampus to hypoxic-ischemic injury (Barnes-Powell, 2007).
Iron deficiency of the heart, liver and skeletal muscle has been shown to affect cellular energy production and organ performance. Arguably, sever iron deficiency may lead to similar deficits in cellular energy metabolism and organ performance in the brain, resulting in a reduced ability to respond to restriction of oxygen and perfusion and in greater hippocampus damage. The hippocampus, especially the DG, is one of the iron rich areas of the rat brain, finding suggest that the integrity of the prenatal hippocampus and its cholinergic input are important for normal development of memory and learning (Ryan and Geckle, 2000). In other hand, this area of the brain (hippocampus) particularly the dentate region is also vulnerable to damage when glucocorticoids are elevated as occurs, for example, when an organism is stressed (Gispen and Biessels, 2000).
Uncontrolled experimental diabetes induced by (STZ) in rats is an endogenous chronic stressor that produces retraction and simplification of apical dendrites of hippocampal CA3 pyramidal neurons (Damasceno et al., 2002).
One effect, synaptic vesicle depletion and dendrite atrophy occurs in diabetes as well as after repeated stress and cort treatment. These changes occurred in concert with adrenal hypertrophy and elevated basal cort release as well as hypersensitivity and defective shut off of cort secretion after stress. Thus as an endogenous stressor STZ diabetes not only accelerates the effects of exogenous stress to alter hippocampal morphology: it also produce structural changes that overlap only partially with those produced by stress and cort in the nondiabetic state (Magariños and McEwen, 2000).
The hippocampal morphological changes induced by stress are mediated by interactions between Gc secretion, excitatory amino acid and are also correlated with deficits in hippocampal dependent memory (DeBoer et al., 2005).
These results and present results confirm that an oxidative imbalance occurs in the hippocampus of neonates born from diabetic rats as we have earlier shown in diabetic rats. Several studies have pointed out that NF-kB (nuclar factor) activation is inhibited by a variety of antioxidants, such as N-acetyl-cystein, butylanted hydroxyl anisole, vitamin E and lipoic acid (Aragno et al., 2000). These data suggest that antioxidants effect some steps of signaling events leading to phosphorylation, ubiquination and degradation (Patil et al., 2006). The role of oxidative stress and Nk-kB activation on diabetic complications is well documented, moreover antioxidant treatment exerts a beneficial effect in experimental models of chronic injury in diabetes and treatment with antioxidants can significantly reduce diabetic complications (Magariños and McEwen, 2000). Reactive oxygen species activates a variety of target genes linked to the development of diabetic complications (Rakeshwar et al., 2006).
In addition, the loss of arachidonic acid content of the synaptosomal membrane, induced by diabetes and by transient cerebral ischemia, making the membrane more resistant to oxidative stress. Oxidative stress induced by chronic hyperglycemia directly can damage ionic homeostasis and membrane transport systems in the brain (Aragno et al., 2000) and may be this is one of the reasons for hippocampal neurons death. Apoptosis in diabetes has been ascribed to hyperglycemia and oxidative stress (Mumby et al., 2002).
However, other experimental studies on streptozotocin induced rat diabetic, showed pathological changes, such as so-called dark neurons and neuronal loss, in different cerebral regions, especially in the hippocampus. A dominant opinion is that hyperglycemia aggravates ischaemic brain damage in experimental STZ-diabetes with transient cerebral ischemia in rats (Piotrowski et al., 2001). It has been suggested that diabetes and ischaemia evoke the oxidative stress following an impairment of the respiratory chain in mitochondria and an overproduction of the Reactive Oxygen Species (ROS). ROS are considered as a main factor in the pathogenesis on neuronal death (Piotrowski et al., 2001).
The other reason for neuronal death in diabetic pregnancy is Iscemia. It has been postulated that neurons with higher oxygen consumption in normal conditions are more susceptible to ischemia insult (Ristow, 2004).
Some studies shown that there is differences in the neural death between diabetic and Ischaemia (Piotrowski et al., 2001) that differences in the neuronal death pathomechanism among hippocampal sectors in diabetes and ischaemia may be related to the different duration and intensity of the oxidative stress in both applied models (acute in ischaemia and chronics in STZ-induced diabetes). It has been postulated that in cerebral ischaemia neuronal apoptosis may be followed by necrotic phase (Patil et al., 2006). Pathomechanism of degenerative changes and neuronal loss through apoptosis or necrosis is not clear until now. It has been suggested that changes in intracellular calcium concentrations in oxidative stress may indicate the pathway of cell death. It is suggested that more sever injury with high intracellular calcium concentration (Ca+2) promotes necrotic cell death, where low (Ca+2) and milder injury promotes cell death through apoptosis (Misumi et al., 2008). Studies on antioxidative treatment would deliver further data important in the exploration of neuronal death in diabetes and ischemia.
In total, it is concluded that maternal diabetes induces some changes in hippocampus neuronal structure and density. Statistical analysis show significant decrease in neuronal density (ND) of CA3 in neonates from diabetic mothers compare to control. Although ND in CA1, CA2 have showed decrease but it is not meaningful. Maybe neurogenic process in hippocampus is responsible for these changes. Therefore, it is better in diabetic pregnant mother the level of glucose was maintained under normal condition.
REFERENCES
- Aragno, M., S. Parola, E. Brignardello, A. Mauro and E. Tamagno et al., 2000. Dehydroepiandrosterone prevents oxidative injury induced by transient ischemia/reperfusion in the brain of diabetic rats. Diabetes, 49: 1924-1931.
PubMed - Aragno, M., R. Mastrocola, E. Brignardello, M. Catalano and G. Robino et al., 2002. Dehydroepiandrosterone modulates nuclear factor-kappaB activation in hippocampus of diabetic rats. Endocrinology, 143: 3250-3258.
PubMed - Artola, A., 2008. Diabetes, stress and ageing-related changes in synaptic plasticity in hippocampus and neocortex the same metaplastic process? Eur. J. Pharmacol., 585: 153-162.
PubMed - Arvanitakis, Z., R.S. Wilson, J.L. Bienias, D.A. Evans and D.A. Bennett, 2005. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch. Neurol., 62: 330-330.
PubMed - Barnes-Powell, L.L., 2007. Infants of diabetic mothers: The effects of hyperglycemia on the fetus and neonate. Neonatal Network, 26: 283-290.
PubMed - Damasceno, D.C., G.T. Volpato, P.C.I de Mattos and R.MV. Cunha, 2002. Oxidative stress and diabetes in pregnant rats. Anim. Reprod. Sci., 15: 235-244.
PubMed - DeBoer, T., S. Wewerka, P.J. Bauer, M.K. Georgieff and C.A. Nelson, 2005. Explicit memory performance in infants of diabetic mothers at 1 year of age. Dev. Med. Child Neurol., 47: 525-531.
PubMed - Deregnier, R.A., C.A. Nelson, K.M. Thomas, S. Wewerka and M.K. Georgieff, 2000. Neurophysiologic evaluation of auditory recognition memory in healthy newborn infants and infants of diabetic mothers. J. Pediatr., 137: 777-784.
PubMed - Gao, Q. and Y.M. Gao, 2007. Hyperglycemic condition disturbs the proliferation and cell death of neural progenitors in mouse embryonic spinal cord. Int. J. Dev. Neurosci., 25: 349-357.
PubMed - Gasparini, L. and H. Xu, 2003. Potential roles of insulin and IGF-1 in Alzheimer's disease. Trends Neurosci., 26: 404-406.
Direct Link - Gispen, W.H. and G.J. Biessels, 2000. Cognition and synaptic plasticity in diabetes mellitus. Trends Neurosci., 23: 542-549.
PubMedDirect Link - Gundersen, H.J.G., T.F. Bendtsen, L. Korbo, N. Marcussen and A. Moller et al., 1988. Some new, simple and efficient stereological methods and their use in pathological research and diagnosis. APMIS., 96: 379-394.
CrossRefPubMedDirect Link - Kinney, B.A., M.B. Rabe, R.A. Jensen and R.W. Steger, 2003. Maternal hyperglycemia leads to gender-dependent deficits in learning and memory in offspring. Exp. Biol. Med., 228: 152-159.
PubMed - Luo, Y., C. Kaur and E.A. Ling, 2002. Neuronal and glial response in the rat hypothalamus-neurohypophysis complex with streptozotocin-induced diabetes. Brain Res., 925: 42-54.
CrossRefPubMedDirect Link - Magariños, A.M. and B.S. McEwen, 2000. Experimental diabetes in rats causes hippocampal dendritic and synaptic reorganization and increased glucocorticoid reactivity to stress. Proc. Natl. Acad. Sci. USA., 97: 11056-11061.
PubMed - Misumi, Y., T. Yamato, T. Obata and M. Aomine , 2008. Effects of ion channel blockers on basal hippocampal monoamine levels in freely moving diabetic and non-diabetic rats. Int. J. Neurosci., 118: 761-780.
PubMed - Mumby, D.G., S. Gaskin, M.J. Glenn, T.E. Schramek and H. Lehmann, 2002. Hippocampal damage and exploratory Preferences in rats: Memory for objects, places and contexts. Learn Mem., 9: 49-57.
PubMed - Nazer, J. and R. Ramírez, 2000. Congenital malformations in the offspring of diabetic mothers. Rev. Med. Child., 128: 1045-1052.
PubMed - Nold, J.L. and M.K. Georgieff, 2004. Infants of diabetic mothers. Pediatr. Clin. North Am., 51: 619-637.
CrossRefPubMedDirect Link - Ornoy, A., 2007. Embryonic oxidative stress as a mechanism of teratogenesis with special emphasis on diabetic embryopathy. Reprod. Toxicol., 24: 31-41.
PubMed - Piotrowski, P., K. Wierzbicka and M. Smiałek, 2001. Neuronal death in the rat hippocampus in experimental diabetes and cerebral ischaemia treated withantioxidants. Folia Neuropathol., 39: 147-154.
PubMed - Polanco, P.A.C., M.C.R. Monsalve, M.A.P. Garibay and A.S. Islas, 2005. Effect of maternal diabetes on human and rat fetal development. Ginecol. Obstet. Mex., 73: 544-552.
PubMed - Rakeshwar, S. Guleria, J. Pan, D. Dipette and U.S. Singh, 2006. Hyperglycemia inhibits retinoic acid induced activation of rac1, prevents differentiation of cortical neurons and causes oxidative stress in a rat model of diabetic pregnancy. Diabetes, 55: 3326-3334.
CrossRef - Ristow, M., 2004. Neurodegenerative disorders associated with diabetes mellitus. J. Mol. Med., 82: 510-529.
PubMed - Ryan, C.M. and M. Geckle, 2000. Why is learning and memory dysfunction in type 2 diabetes limited to older adults? Diabet. Metab. Res. Rev., 16: 308-315.
PubMed - Sima, A.A. and Z.G. Li, 2005. The effect of C-peptide on cognitive dysfunction and hippocampal apoptosis in type 1 diabetic rats. Diabetes, 54: 1497-1505.
PubMed - Sinclair, A.J., A.J. Girling and A.J. Bayesr, 2000. Cognitivedysfunction in older subjects with diabetes mellitus: Impact on diabetes self-management and use of care services. All Wales Research into Elderly (AWARE) Study. Diabetes Res, Clin. Pract., 50: 203-212.
PubMed