
Emmanuel Edet Etim
Department of Chemical Sciences, Federal University Wukari, PMB 1020, Wukari, Taraba State, Nigeria
LiveDNA: 234.8332
Chrysanthus Andrew
Department of Chemical Sciences, Federal University Wukari, PMB 1020, Wukari, Taraba State, Nigeria
Usman Lawal
Department of Chemical Sciences, Federal University Wukari, PMB 1020, Wukari, Taraba State, Nigeria
Ifeoma Sandra Udegbunam
Department of Chemical Sciences, Federal University Wukari, PMB 1020, Wukari, Taraba State, Nigeria
Etiowo George Ukpong
Department of Science Technology, Akwa Ibom State Polytechnic, Ikot Ekpene
Journal of Applied Sciences
Year: 2020 | Volume: 20 | Issue: 1 | Page No.: 26-34







ABSTRACT
Background and Objectives: Computational studies have gained popularity nowadays as a means of studying simple and complex chemical systems with a view to complement experimental techniques. Hence the objective of this work was to use different computational methods to study the protonation of carbonyl sulphide, its possible protonated analogues and determine the best site for its protonation which will correspond with the experimentally determined proton affinity. Materials and Methods: In this study, quantum chemical calculations were carried out using GAUSSIAN 09 suite of programs using 5 different levels of theory and two basis sets. Bond lengths and angles, vibrational frequencies, dipole moments and proton affinity (PA) are among parameters of interest that have been calculated for OCS and its protonated analogues. In other to evaluate the best computational method, the difference between the calculated value and the reported experimental data is used as a benchmark. Results: CCSD method along with 6-311++G** gives the best prediction for the bond length in the neutral molecule. G4 predicts best the rotational constants and dipole moments for HOCS+ and OCS respectively. The proton affinity values calculated in this work reveals that the protonation of OCS is more favoured via sulphur atom than via oxygen atom. Thus proton affinity corresponds to the most stable protonated analogue with the proton attached to the site of less electron density hence electronegativity is not a good criterion for predicting the protonation site of a molecule. Conclusion: Optimization of OCS molecule and its protonated analogues was performed. We have shown that the protonation of a molecule can be determined by considering electron density rather than electronegativity.
PDF Abstract XML References Citation
Copyright: © 2020. This is an open access article distributed under the terms of the creative commons attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
How to cite this article
Emmanuel Edet Etim, Chrysanthus Andrew, Usman Lawal, Ifeoma Sandra Udegbunam and Etiowo George Ukpong, 2020. Protonation of Carbonyl Sulfide: Ab initio Study. Journal of Applied Sciences, 20: 26-34.
DOI: 10.3923/jas.2020.26.34
URL: https://scialert.net/abstract/?doi=jas.2020.26.34
DOI: 10.3923/jas.2020.26.34
URL: https://scialert.net/abstract/?doi=jas.2020.26.34
INTRODUCTION
A substantial (>200) number of molecular species have been observed from the interstellar and circumstellar media with about 10% being cations and almost 80% of these cations are protonated molecules1-8. Of all the known interstellar molecules, hydrogen predominates the interstellar medium and this implies that all the neutral molecular species present in the ISM are susceptible to protonation. Studies have shown that both the protonated species and their neutral analogues consist part of the interstellar media where the protonated species form as the natural precursors to the neutral species9-10 and this implies that for every neutral species existing in the ISM, there also exist its protonated analogue11-12. Carbonyl sulfide (OCS) is a well known interstellar molecule that was first detected within the giant molecular Sagittarius B2 by Jefferts et al.13 and was further confirmed in about ten interstellar sources14-16. OCS is among the 15 sulfur-containing compounds that have been known from interstellar observations17. This interstellar molecule OCS plays an important role in the global cycling of sulphur18 and due to its high abundance (about 500 ppt) in the troposphere; it forms the major source of stratospheric aerosol19.
Protonations are key constituents of interstellar molecules and are essential to many chemical processes occurring in the ISM. The protonation of a neutral molecule can occur in more than one position and can affect electronic, molecular structure and proton binding energies. The presence of protonated OCS has been postulated in the interstellar clouds15,20. In principle, the OCS molecule has three possible protonation sites (either through O, C or S) which could results in three isomers namely, HOCS+, OC(H)S+ and HSCO+ respectively. Two of these isomers (HOCS+ and HSCO+) are more stable and have been a subject of study21. The HOCS+ isomer was first reported by Nakanaga and Amano22 using a high-resolution infrared spectrum and the authors reported that the vibrational frequency of the O-H stretching mode was 3435.16 cm–1. For the HSCO+ corresponding isomer, these authors were unable to identify the S-H stretching mode. In another experimental study, the rotational spectrum of HOCS+ was observed by Ohshima and Endo23 and the values of their rotational constants were in excellent agreement with the previous work of Nakanaga and Amano22. Though, Ohshima and Endo23 failed to measure the spectrum of HSCO+. Another theoretical and experimental study was conducted by Saebo et al.24, in other to resolve the disagreement between ab Initio and experimental studies of the HSCO+ isomer. Theoretical studies predicted the S-H stretching mode of HSCO+ at 2496 cm–1 and the spectroscopic study was only able to observe HOCS+ but failed to detect the IR absorption of HSCO+. Though, these authors suggested that the reason for not observing HSCO+ was attributed to the low amount which was below the detection limit. Further work from Xia et al.25 with that of McCarthy and Thaddeus26 could not locate the S-H stretching band for the HSCO+. However, in a recent study by Tsuge and Lee21, the IR spectrum of HSCO+ was studied together with HOCS+ and t-HOCS through electron bombardment of a mixture of OCS and para-hydrogen. For the first time, the conflict between theoretical and experimental study for the IR spectra of HSCO+ (v1 = 2506.9, v2 = 2074.2 cm–1) was resolved and new modes (v2 and v3) for HOCS+ was reported.
Motivated by the recent advancement in quantum chemical calculation methods, the present work is focused at determining the proton affinity (PA), vibrational frequencies, rotational constants and dipole moments for OCS and all its possible protonated analogues using five different theoretical methods (HF, B3LYP, MP2, CCSD, G4) with two basis sets (6-311++G** and cc-pVDZ). The differences between the calculated parameters of interest and the corresponding experimental data (where available) have been used to ascertain the efficiency and accuracy of the different computational methods applied. Specifically, the purpose this work was to use results of the different computational method to determine the best site for the protonation of OCS which will correspond with the experimentally determined proton affinity.
MATERIALS AND METHODS
Study area: This research project was conducted from August, 2015 to September, 2016 at the Indian Institute of Science (IISc) Banglore.
Computational procedure: All the quantum chemical calculations reported in this study have been carried out using the GAUSSIAN 09 suit of programs27. In order to obtain highly accurate values for all the parameters investigated in this study for all the molecular species considered, different high-level ab initio methods have been employed. These include the Hartree-Fock (HF), B3LYP, second-order Møller-Plesset perturbation theory (MP2) and coupled-cluster theory (CCSD) with 6-311++G** basis set. Besides this basis set, the Dunning correlation-consistent polarized valence double zeta (cc-pVDZ) basis set which is designed to converge smoothly towards the complete basis set limit is also used with the MP2 method. In addition to the above methods, the Gaussian 4 theory (G4) compound model is also utilized in predicting accurate parameters for all the systems under consideration in this study. This compound model offers high accuracy results at a less computational cost. It consists of different component calculations whose results are then combined in a predefined manner28-36. The choice of these methods and basis sets is based on our previous studies by Etim and Arunan6-7 and Etim et al.8. The OCS molecular species and its possible protonated analogues were optimized with the different methods mentioned above. For the calculation of the proton affinity, only stable equilibrium structures are considered. This was verified via harmonic frequencies calculations with the equilibrium geometries having only real frequencies with no imaginary frequencies. Proton affinity is calculated as the difference in energy (electronic energy) between a neutral species and its protonated analogue. Zero-point correction to energies is included in all the calculations reported here.
Statistical analysis: Standard deviation analysis was used to estimate the error difference between our computational results and the reported experimental values.
RESULTS AND DISCUSSION
To get the best computational method, the difference between the computed parameters of interest and the observed experimental values where available was used as standard. The smaller the error is, the more accurate the computational method would be in predicting the experimental data.
Optimized geometry: Optimized geometries of the neutral OCS and its protonated analogues calculated using 5 different computational methods are presented in Table 1. The geometrical parameters for the bond length and angles for the neutral molecule at the levels of theory used agree well with the observed experimental data reported by Callomon et al.37. A close look at Table 1 reveals that at the same level of theory, the HOC bond angle in HOCS+ is greater than the HSC bond angle in HSCO+. In the HOCS+ analogue, hydrogen is bound to sp2-hybridized oxygen while in the HSCO+ system, hydrogen is bound to a sulfur atom which undergoes hybridization to a lesser degree than in oxygen. In addition, the H-O and H-S bond lengths in the two protonated analogues are quite different with the former been smaller than the later; this can be explained based on hybridization. Upon protonation, it is clear that the C-S bond distance is generally much longer than in the neutral molecule while the C-O distance in the HSCO+ is shorter. Thus, there is a contraction of C-O and elongation of C-S bond lengths in HSCO+ compared to that in the neutral molecule (OCS). On the other hand, the C-S distance in HOCS+ is shorter whereas the C-O bond distance is longer when compared to the neutral molecule. In general, our geometrical calculated values compare favourably with that reported by Saebo et al.24. Furthermore, the protonation of OCS through the oxygen atom results in HOC bond angles of about 120° and through sulfur atom the HSC bond angle is about 90°. The rotational constants for the optimized structures of OCS and two of its protonated analogues at different computational methods used are listed in Table 2.
| Table 1: | Equilibrium geometries of OCS and its protonated analogues |
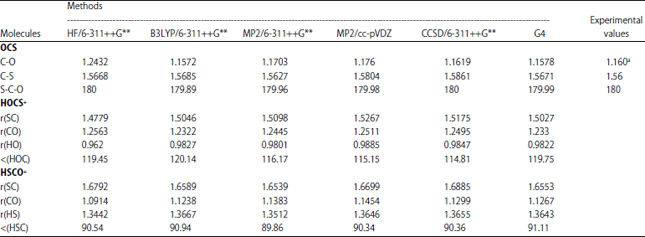 | |
aCallomon et al.37, HF: Hartree-Fock, B3LYP: Becke, 3-parameter, Lee-Yang-Parr, MP2: Second-order Møller-Plesset perturbation theory, CCSD: Coupled-cluster theory, G4: Gaussian 4 theory | |
| Table 2: | Rotational constants (GHz) for protonated OCS |
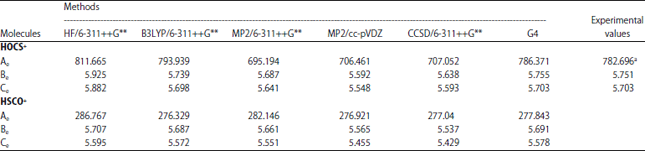 | |
aSaebo et al.24, HF: Hartree-Fock, B3LYP: Becke, 3-parameter, Lee-Yang-Parr, MP2: Second-order Møller-Plesset perturbation theory, CCSD: Coupled-cluster theory, G4: Gaussian 4 theory, Ae, Be, Ce: Rotational constants | |
| Table 3: | Dipole moments for OCS and its protonated analogues |
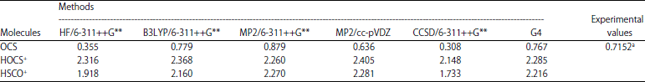 | |
aLahaye et al.39, HF: Hartree-Fock, B3LYP: Becke, 3-parameter, Lee-Yang-Parr, MP2: Second-order Møller-Plesset perturbation theory, CCSD: Coupled-cluster theory, G4: Gaussian 4 theory | |
Based on the different levels of theory used, the G4 level agrees excellently well with a difference of about 3.675 GHz from the experimental values of HOCS+ reported by Nakanaga and Amano22. The largest deviation between the ab initio used and the observed experimental rotational constants is found at the Hartree-Fock level of theory.
A general look at the rotational constants for HOCS+ in this work indicates that the values are well predicted even though the results of the computational methods used are based on equilibrium structures while the experimental data are obtained from vibrationally averaged structures. The rotational constants of HSCO+ have not been determined experimentally and we have compared our results with other theoretical results. Our rotational constants using MP2 at 6-311++G** basis set and G4 methods are in excellent agreement with the theoretical values reported by Saebo et al.24 and Wheeler et al.38. A comparison of the different computational methods used in calculating the dipole moments (Table 3) indicates that the dipole moment of OCS is well predicted using G4 level with a difference of only 0.05 from the experimental value of Lahaye et al.39.
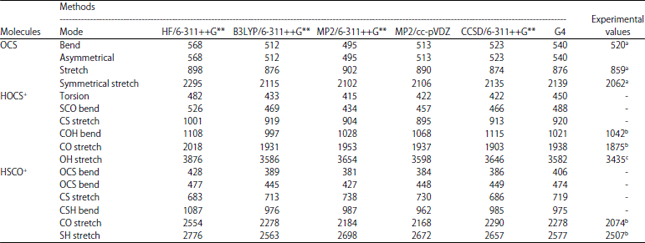
IR spectra: The vibrational wave-numbers of the two protonated carbonyl sulfide (HSCO+ and HOCS+) have been widely studied both experimentally and with different computational techniques. The results of the predicted harmonic vibrational frequencies of OCS and its protonated analogues (HSCO+ and HOCS+) using different ab initio methods are presented in Table 4. Figure 1a-f depicts the IR spectra of the OCS at different levels of theories and Fig. 2a-f represents the corresponding IR spectra of the protonated analogues. For OCS which is a linear molecule, there will be 3N-5 modes of vibrations40. Hence only four vibrational modes are possible which include; symmetrical stretching (v1), asymmetrical stretching (v3) and two of the degenerated bending modes (v2). Comparing the five computational methods used in this work reveals that the HF method gave a larger average error (8.5%) between the calculated and experimental frequencies and MP2 at 6-311++G** basis set predicted the best frequencies with an average percentage error of 1.66%. Generally, the HF method is known with the fact that it does not take into account electron correlation which often overestimates the vibrational frequencies by about 10%41. On the basis of a specific frequency, the CCSD method predicted the best bending vibrational frequency followed by MP2 and B3LYP were the differences between the calculated frequencies and the observed experimental value is very small. On the other hand, the two protonated analogues (HOCS+ and HSCO+) are all non-linear and will have 3N - 6 vibrations giving rise to six vibrational frequencies. For the HOCS+, only three of these frequencies (O-H stretch, C-O stretch, HOC bend) have been observed experimentally21-22. Our predicted vibrational frequency for the O-H stretch (3582 cm–1) at the G4 level of theory (without empirical corrections or scaling) was the method with the least error (147 cm–1) when compared with the experimental data of Nakanaga and Amano22 and other computational methods.
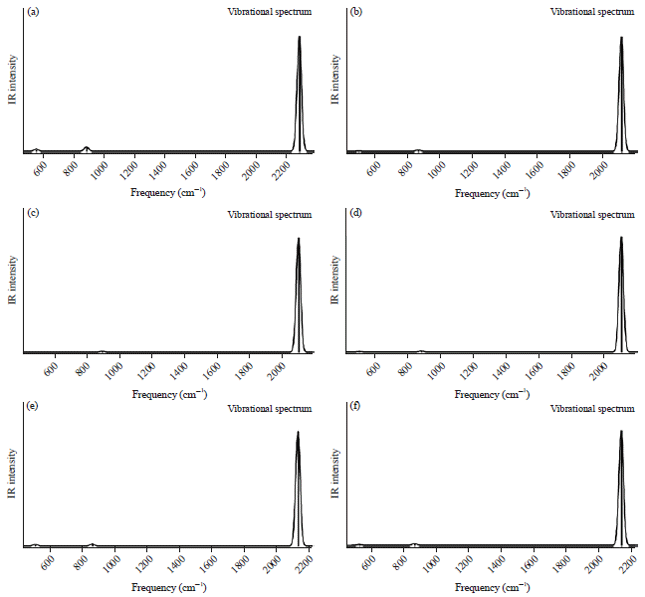 | |
| Fig. 1(a-f): | IR spectra of SCO at (a) HF/6-311++G**, (b) B3LYP/6-311++G**, (c) MP2/6-311++G**, (d) MP2/cc-pVDZ, (e) CCSD//6-311++G** and (f) G4 level |
| Table 4: | Vibrational frequencies (cm–1) for OCS and its protonated analogues |
 | |
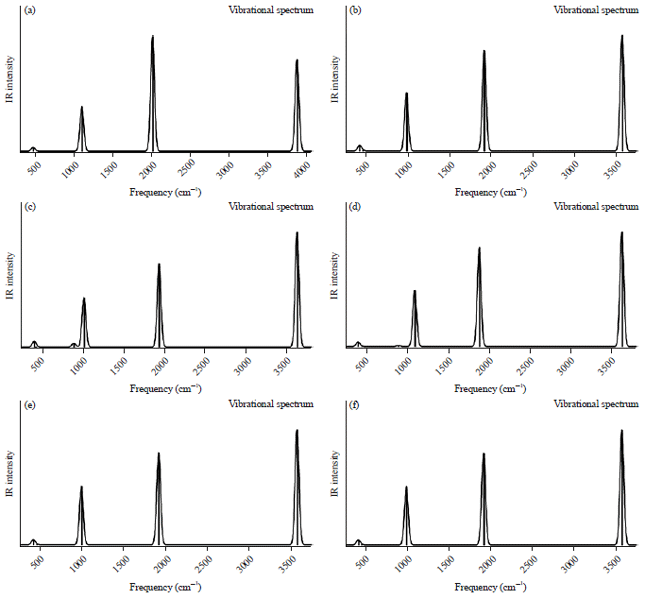 | |
| Fig. 2(a-f): | IR spectra of protonated SCO at (a) HF/6-311++G**, (b) B3LYP/6-311++G**, (c) MP2/6-311++G**, (d) MP2/cc-pVDZ, (e) CCSD/6-311++G** and (f) G4 level |
The G4 method also produced the best result for the predicted HOC bend frequency for the HOCS+ isomer. The C-O stretch frequency was much accurately predicted at the CCSD level of theory. The H-S vibrational frequency for HSCO+ protonated analogue has been a subject of experimental observation not until recently where Tsuge and Lee21 observed the S-H frequency at 2507 cm–1. For the H-S vibrational frequency, 2 methods; B3LYP and G4 computed the accurate frequency with an error of 56 and 70 cm–1 respectively when compared with other methods. The C-O stretch frequency was better predicted at the MP2 level with the cc-pVDZ basis set.
Proton affinity (PA): Table 5 reports the proton affinities for the protonation of OCS calculated at different levels of theory together with the corresponding errors. Figure 3 shows the corresponding optimized structures of protonated analogues for OCS. It is clear from Table 5 that the results of the five computational methods used in calculating the PA indicates that the PA is better predicted when protonation occurs via sulfur atom (i.e., a site with less electron density) than at oxygen atom (a site with high electron density) even though oxygen is more electronegative than sulfur. Therefore, PA corresponds to the most stable protonated analogue with the proton attached to the site of less electron density.
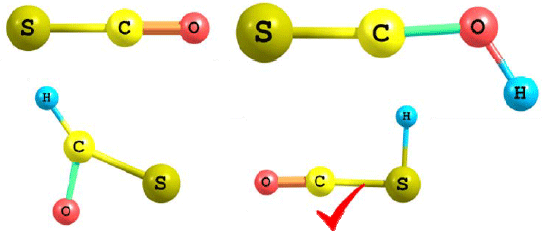 | |
| Fig. 3: | Optimized structures of protonated analogues of OCS |
Source: Gaussian-09 suit program | |
| Table 5: | Proton affinity (PA) of OCS at different sites of protonation |
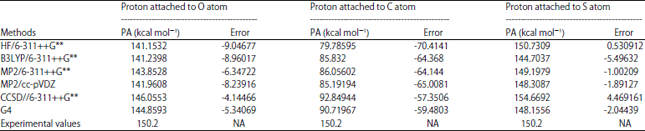 | |
HF: Hartree-Fock, B3LYP: Becke, 3-parameter, Lee-Yang-Parr, MP2: Second-order Møller-Plesset perturbation theory, CCSD: Coupled-cluster theory, G4: Gaussian 4 theory, NA: Not applicable | |
Hence, electro-negativity of an atom is not a good criterion for deciding the site of protonation. Thus, from the calculated PA in this work, S-protonation is more favoured than O-protonation which indicates that HSCO+ is more stable than HOCS+. This also confirms the previously reported ab initio studies that HSCO+ protonated form is more stable than HOCS+24,38,42-45.
CONCLUSION
Protonation of carbonyl sulfide was studied through ab initio methods to ascertain the best computational method by determining the difference between the computed and experimental values. The five computational methods used in calculating the PA indicate that the PA is better predicted when protonation occurs via sulfur atom i.e., a site with less electron density than at oxygen atom, a site with high electron density even though oxygen is more electronegative than sulfur. Generally, CCSD and G4 methods give the best prediction of the parameters of interest calculated. This study has shown that electronegativity is not the only criterion when predicting the protonation of a molecule.
SIGNIFICANCE STATEMENT
This study discovered that the protonation of OCS is more favoured via sulphur atom than via oxygen atom. Thus proton affinity corresponds to the most stable protonated analogue with the proton attached to the site of less electron density hence electronegativity is not a good criterion for predicting the protonation site of a molecule.
ACKNOWLEDGMENTS
EEE acknowledges a research fellowship from the Indian Institute of Science, Bangalore. Authors acknowledge the Computational Research Facilities of IISc Bangalore.
REFERENCES
- Fraser, H.J., M.R.S. McCoustra and D.A. Williams, 2002. Astrochemistry: The molecular universe. Astron. Geophys., 43: 2.10-2.18.
CrossRefDirect Link - Tielens, A.G.G.M., 2013. The molecular universe. Rev. Mod. Phys., Vol. 85, No. 3.
CrossRefDirect Link - Etim, E.E. and E. Arunan, 2015. Rotational spectroscopy and interstellar molecules. Planex Newslett., 5: 16-21.
Direct Link - Etim, E.E., P. Gorai, A. Das, S.K. Chakrabarti and E. Arunan, 2016. Systematic theoretical study on the interstellar carbon chain molecules. Astrophys. J., Vol. 832, No. 2.
CrossRefDirect Link - Etim, E.E. and E. Arunan, 2016. Interstellar isomeric species: Energy, stability and abundance relationship. Eur. Phys. J. Plus, Vol. 131.
CrossRef - Etim, E.E. and E. Arunan, 2017. Partition function and astronomical observation of interstellar isomers: Is there a link? Adv. Space Res., 59: 1161-1171.
CrossRefDirect Link - Etim, E.E. and E. Arunan, 2017. Accurate rotational constants for linear interstellar carbon chains: Achieving experimental accuracy. Astrophys. Space Sci., Vol. 362.
CrossRefDirect Link - Etim, E.E., P. Gorai, A. Das, and E. Arunan, 2017. C5H9N Isomers: Pointers to possible branched chain interstellar molecules. Eur. Phys. J., Vol. 71.
CrossRefDirect Link - DeFrees, D.J. and A.D. McLean, 1986. A priori predictions of the rotational constants for protonated formaldehyde and protonated methanol. Chem. Phys. Lett., 131: 403-408.
CrossRefDirect Link - Cheikh, F., F. Pauzat and Y. Ellinger, 2003. A theoretical study of protonated OCnOH+ ions, n=3-8. Chem. Phys., 290: 147-162.
CrossRefDirect Link - Herbst, E., 2005. Molecular ions in interstellar reaction networks. J. Phys.: Conf. Ser., 4: 17-25.
CrossRefDirect Link - Jefferts, K.B., A.A. Penzias, R.W. Wilson and P.M. Solomon, 1971. Detection of interstellar carbonyl sulfide. Astrophys. J., 168: L111-L113.
Direct Link - Goldsmith, P.F. and R.K. Linke, 1981. A study of interstellar carbonyl sulfide. Astrophys. J., 245: 482-494.
CrossRefDirect Link - Fock, W. and T. McAllister, 1982. Probable abundance ratios for interstellar HCS2+, HCOS, HCO2+. Astrophys. J., 257: L99-L101.
Direct Link - Palumbo, M.E., T.R. Geballe and A.G. Tielens, 1997. Solid carbonyl sulfide (OCS) in dense molecular clouds. Astrophys. J., 479: 839-844.
CrossRefDirect Link - Lovas, F.J., 2004. NIST recommended rest frequencies for observed interstellar molecular microwave transitions-2002 revision. J. Phys. Chem. Reference Data, 33: 177-355.
CrossRefDirect Link - Thornton, D.C., A.R. Bandy, B.W. Blomquist and B.E. Anderson, 1996. Impact of anthropogenic and biogenic sources and sinks on carbonyl sulfide in the North Pacific troposphere. J. Geophys. Res.: Atmos., 101: 1873-1881.
CrossRefDirect Link - Hiraoka, K., K. Fujita, M. Ishida, K. Hiizumi and F. Nakagawa et al., 2005. Thermochemical stabilities and structures of the cluster ions OCS+, S2+, H+(OCS) and C2H5+ with OCS molecules in the gas phase. J. Am. Soc. Mass Spectrom., 16: 1760-1771.
CrossRefDirect Link - Turner, B.E., T. Amano and P.A. Feldman, 1990. Searches for the protonated interstellar species HC3NH+, CH3CNH+ and HOCS+: Implications for ion-molecule chemistry. Astrophys. J., 349: 376-387.
Direct Link - Tsuge, M. and Y.P. Lee, 2016. Infrared spectra of two isomers of protonated carbonyl sulfide (HOCS+ and HSCO+) and t-HOCS in solid para-hydrogen. J. Chem. Phys., Vol. 145, No. 16.
CrossRefDirect Link - Nakanaga, T. and T. Amano, 1987. High-resolution infrared identification of HOCS+ with difference frequency laser spectroscopy. Mol. Phys., 61: 313-323.
CrossRefDirect Link - Ohshima, Y. and Y. Endo, 1996. Rotational spectroscopy of jet-cooled molecular ions and ion complexes. Chem. Phys. Lett., 256: 635-640.
CrossRefDirect Link - Saebo, S., M.M. Sanz and S.C. Foster, 1997. Structures and vibrational spectra for protonated carbonyl sulfide. Theoret. Chem. Accounts, 97: 271-276.
CrossRefDirect Link - Xia, C., M.M. Sanz and S.C. Foster, 1998. Diode laser spectroscopy of the ν1 and ν3 bands of SD+3. J. Mol. Spectrosc., 188: 175-181.
CrossRefDirect Link - McCarthy, M.C. and P. Thaddeus, 2007. Laboratory detection of the elusive HSCO+ isomer. J. Chem. Phys., Vol. 127, No. 22.
CrossRefDirect Link - Moller, C. and M.S. Plesset, 1934. Note on an approximation treatment for many-electron systems. Phys. Rev., 46: 618-622.
CrossRefDirect Link - Pople, J.A., R. Krishnan, H.B. Schlegel and J.S. Binkley, 1978. Electron correlation theories and their application to the study of simple reaction potential surfaces. Int. J. Quantum Chem., 14: 545-560.
CrossRefDirect Link - Scuseria, G.E., C.L. Janssen and H.F. Schaefer III, 1988. An efficient reformulation of the closed-shell coupled cluster single and double excitation (CCSD) equations. J. Chem. Phys., 89: 7382-7387.
CrossRefDirect Link - Dunning, Jr. T.H., 1989. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys., 90: 1007-1023.
CrossRefDirect Link - Martin, J.M.L. and G. de Oliveira, 1999. Towards standard methods for benchmark quality ab initio thermochemistry-W1 and W2 theory. J. Chem. Phys., 111: 1843-1856.
CrossRefDirect Link - Parthiban, S. and J.M. Martin, 2001. Assessment of W1 and W2 theories for the computation of electron affinities, ionization potentials, heats of formation and proton affinities. J. Chem. Phys., 114: 6014-6029.
CrossRefDirect Link - Curtiss, L.A., K. Raghavachari, P.C. Redfern, V. Rassolov and J.A. Pople, 1998. Gaussian-3 (G3) theory for molecules containing first and second-row atoms. J. Chem. Phys., 109: 7764-7776.
CrossRefDirect Link - Curtiss, L.A., P.C. Redfern and K. Raghavachari, 2007. Gaussian-4 theory. J. Chem. Phys., Vol. 126, No. 8.
CrossRefDirect Link - Curtiss, L.A., P.C. Redfern and K. Raghavachari, 2007. Gaussian-4 theory using reduced order perturbation theory. J. Chem. Phys., Vol. 127, No. 12.
CrossRefDirect Link - Wheeler, S.E., Y. Yamaguchi and H.F. Schaefer III, 2006. Protonated carbonyl sulfide: Prospects for the spectroscopic observation of the elusive HSCO+ isomer. J. Chem. Phys., Vol. 124, No. 4.
CrossRefDirect Link - Lahaye, J.G., R. Vandenhaute and A. Fayt, 1987. CO2 laser saturation Stark spectra and global rovibrational analysis of the main isotopic species of carbonyl sulfide (OC34S, O13CS and 18OCS). J. Mol. Spectrosc., 123: 48-83.
CrossRefDirect Link - Watson, J.K., 1993. Vibration-rotation Hamiltonians of linear molecules. Mol. Phys., 79: 943-951.
CrossRefDirect Link - Wang, Z.D., M. Hysmith, M. Yoshida, B. George and P.C. Quintana, 2014. Theoretical study of the vibrational frequencies of carbon disulfide. Int. J. Quantum Chem., 114: 429-435.
CrossRefDirect Link - Fortenberry, R.C., X. Huang, J.S. Francisco, T.D. Crawford and T.J. Lee, 2012. Fundamental vibrational frequencies and spectroscopic constants of HOCS+, HSCO+ and isotopologues via quartic force fields. J. Phys. Chem. A, 116: 9582-9590.
CrossRefDirect Link - McAllister, T., P.R. Taylor and M. Scarlett, 1986. Thermochemistry of HCOS+ and HCS2+. Org. Mass Spectrom., 21: 157-159.
CrossRefDirect Link - Taylor, P.R. and M. Scarlett, 1985. The rotational spectra of HOCO+, HOCS+, HSCO+ and HSCS+. Astrophys. J., 293: L49-L51.
Direct Link - Liu, J., B. van Devener and S.L. Anderson, 2002. The effects of vibrational mode and collision energy on the reaction of formaldehyde cation with carbonyl sulfide. J. Chem. Phys., 117: 8292-8307.
CrossRefDirect Link