
C.U. Ibeji
Department of Pure and Industrial Chemistry, University of Nigeria, Nsukka, Nigeria
J. Adegboyega
Department of Chemistry, University of Ibadan, Nigeria
O.D. Okagu
Department of Pure and Industrial Chemistry, University of Nigeria, Nsukka, Nigeria
B.B. Adeleke
Department of Chemistry, University of Ibadan, Nigeria
Journal of Applied Sciences
Year: 2016 | Volume: 16 | Issue: 11 | Page No.: 504-516







ABSTRACT
Background: Conjugated systems are of great interest to chemists, particularly photochemists and spectroscopists. Benzophenone (BP), a highly conjugated compound is a well-known compound attracting keen interest from photochemist. Methodology: A comparative knowledge of the nature of ground state of dimethoxythiobenzophenone (DMTB) and dimethoxybenzophenone, their geometries, band gaps, absorption and transition properties will be a valuable contribution for further studies and practical applications. Results: The UV analysis of benzophenone (BP) in two solvents (ethanol, ε = 24.5 and n-hexane, ε = 1.88) and FT-IR correlated with Density Functional Theory (DFT) and Time-Dependent Density Functional Theory (TDDFT) of benzophenone (BP), thiobenzophenone (TBP), dimethoxybenzophenone (DMB) and dimethoxythiobenzophenone (DMTB) suggests a singlet ground state. Conclusion: The DMTB absorbs at the visible region with a lower band gap than DMB. The BP exhibits two bands: (I) A red shift from 247.6 nm (non-polar solvent) to 252.3 (polar solvent), Δλ = +4.7 nm and (II) A blue shift from 205.3 nm (non-polar solvent) to 197.7 nm (polar solvent), Δλ = -7.6 nm. This shows that band I is likely to be π→π* and band II n→π*. The DMB like BP absorbs at the UV region of the spectrum and unlike DMTB whose absorption is principally at the visible region. The DMTB showed a longer bond length of the thiocarbonyl (C=S) than carbonyl (C=O) in BP and DMB.
PDF Abstract XML References Citation
Received: February 23, 2016;
Accepted: September 10, 2016;
Published: October 15, 2016
How to cite this article
C.U. Ibeji, J. Adegboyega, O.D. Okagu and B.B. Adeleke, 2016. Nature of Ground State of Benzophenone and some of its Substituted Derivatives: Experimental and DFT Study. Journal of Applied Sciences, 16: 504-516.
DOI: 10.3923/jas.2016.504.516
URL: https://scialert.net/abstract/?doi=jas.2016.504.516
DOI: 10.3923/jas.2016.504.516
URL: https://scialert.net/abstract/?doi=jas.2016.504.516
INTRODUCTION
The effect of solvent on the UV spectra of substituted benzophenones was studied in order to detect the existence of molecular interactions and determine their UV spectroscopic behaviour1. In benzophenones, both phenyls can interact with the C=O group through σ (inductive effect) and π (mesomeric effect) bonds. The overlapping between the π bonds of the rings and of the C=O form a MO that comprises the entire molecule. Due to this π-electronic delocalization, the C=O group loses part of its individual character and partially integrates with the phenyls, leading to system stabilization and transference of the electronic deficiency from the carbonyl group toward the atoms of the substituents1.
The need to study various derivatives of benzophenone is pressing giving the various novelties resulting there from suitable for various purposes. Comparison of benzophenone with its thio derivative whose only difference from benzophenone (BP) lies in sulphur, which is in the same group VI as oxygen, provides a great basis for an outstanding progress in the chemistry of benzophenone.
Thiobenzophenone is the prototypical thioketone. Unlike other thioketones that tend to dimerize to form rings and polymers, thiobenzophenone is quite stable, although it photo-oxidizes in air to form benzophenone and sulphur2. According to the double bond rule, the C=S double bond of most thioketones is unstable with respect to dimerization making the stability of thiobenzophenone valuable for studying C=S chemistry2. The energy difference between the p orbitals of sulphur and carbon is greater than that between oxygen and carbon in ketones3. The relative difference in energy and atomic orbitals of sulfur compared to carbon results in poor overlap of the orbitals and the energy gap between the HOMO and LUMO is thus reduced for C=S relative to C=O4. A variety of thiones with structures and stability related to thiobenzophenone have also been prepared2.
MATERIALS AND METHODS
Experimental procedure: Benzophenone, diethyl ether, ethanol and n-hexane were obtained from the chemical store, Department of Chemistry, University of Ibadan. The obtained compound was purified by recrystallization in diethyl ether. Diethyl ether was heated to boiling in a flask; an amount sufficient to dissolve the solid was added to the flask containing about 2 g of benzophenone and the flask was swirled gently to dissolve the solid. The flask was placed on hot plate to keep the solution warm and the solution was filtered hot. The flask was left to stand until the crystals were properly formed and the solvent allowed evaporating leaving the pure crystals.
The purity of the compound was ascertained by melting point determination. This was done by the apparatus Barnstead electrothermal, which showed that the compound was pure as it melted between 48.9 and 49.7°C which agrees with literature value of 48.5°C5 giving the laboratory conditions at which the determination was done. Various concentrations of BP were prepared from already prepared stock solutions in ethanol and n-hexane by dilution principle. The stock of 50, 25, 12.5 and 10 ppm were prepared giving a concentration of 2.74×10–4, 1.37×10–4, 6.85×10–5 and 5.49×10–5 M, respectively.
The stock was prepared by dissolving 5 mg of benzophenone in the solvent in a 100 mL standard flask and making it up to the mark.
The UV of the prepared solution was taken by PerkinElmer Spectrum Lambda UV-Vis spectrometer while, the IR of BP was taken using KBr disc with PerkinElmer spectrum version 10.4.3.
Computational method: Quantum mechanical calculations of the ground state molecular structures, dipole moments, polarizabilities, energies and frontier orbital energies (EHOMO and ELUMO), inorder to determine ground state of the systems, singlet-triplet gaps was carried out using the method by Ibeji et al.6. Spectroscopic properties of benzophenone (BP) and three of its derivatives namely thiobenzophenone (TBP), dimethoxybenzophenone (DMB) and dimethoxythiobenzophenone (DMTB) were done using ab-initio restricted HF-DFT self-consistent field (B3LYP) level of theory in vacuum on a 2.40 GHz personal computer. The geometry and energy optimisations of these compounds leading to the energy minima were first performed without any symmetry constraints using the analytical gradient method of B3LYP by means of the standard polarised basis set incorporated in Spartan7. Various conformers were obtained from which the optimised geometries of the best conformers (least energy forms) were obtained; the forms which were eventually used in obtaining the parameters of interest.
Also, Mullikan charges, bonds such as C=C, C=O, C=S, C-H, C-O together with other geometric parameters such as bond lengths and angles, dihedral angles etc. were calculated all calculations were done using Spartan 14 software package8.
RESULTS AND DISCUSSION
Molecular optimisation: Density functional theory conformational analysis for benzophenone (BP), thiobenzophenone (TBP), dimethoxybenzophenone (DMB) and dimethoxythiobenzophenone (DMTB) gave rise to different conformers (Fig. 1). The best conformers i.e., the most stable conformers, obtained using 6-31 G (diffuse) basis set were obtained. The number of conformer and some of the molecular properties of the compounds are presented in Table 1.
Table 1 shows the effect of change in functional group and addition of substituents on the symmetry and conformations of the studied compounds.
Molecular geometry: The three-dimensional arrangement of the compounds were analysed and specified by bond lengths, bond angles and dihedral angles as presented in Table 2.
Difference in the chemistry of S and O are specified from the understanding of O=O double bonds which are much stronger than S=S, S-S single bonds almost twice as strong as O-O single bonds, sulphur (EN = 2.58), much less electronegative than O (EN = 3.44) and ability of sulphur to expand its valence shell to hold more than eight electrons unlike oxygen.
The radius of a sulphur atom is about 60% larger than that of an oxygen atom:
As a result, it is harder for sulphur atoms to come together in forming double bonds with itself or atoms of other elements, therefore double bonds formed by S is much weaker than that formed by O. As a result, bond dissociation enthalpy for a C=S double bond is 477 kJ mol–1 whereas the bond dissociation enthalpy for a C=O double bond is 745 kJ mol–1 (Bodner Research web, the chemistry of oxygen and sulphur).
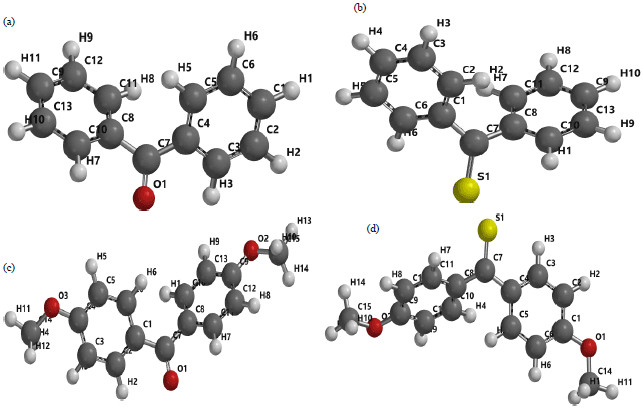 | |
| Fig. 1(a-d): | Optimized structure of (a) BP, (b) TBP, (c) DMB and (d) DMTB |
| Table 1: | Some of the parameters of the studied compounds obtained from their optimisation |
 | |
| Table 2: | Geometric properties of DMB and DMTB |
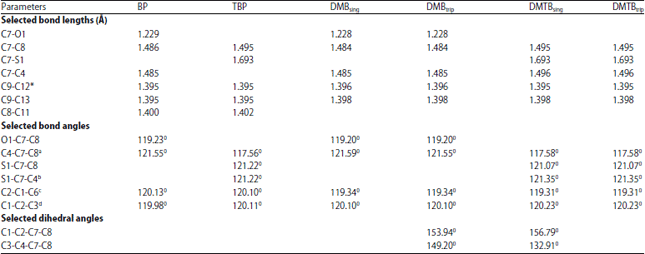 | |
*Used to designate equivalence of C9-C12 to C1-C2, C1-C6 and C9-C13 in BP (C1 and C9 being the para-carbons) and TBP (C3 and C9 being the para-carbons; the bond length labels being C3-C4, C4-C5, C9-C12, C9-C13), aCorresponding label for TBP is C1-C7-C8, bCorresponding label for TBP is S1-C7-C1, cCorresponding label for TBP is C3-C4-C5 and dCorresponding label for TBP is C2-C3-C4 | |
Below are some of the selected geometrical properties of DMB and DMTB obtained using B3LYP/6-31+G (d).
From the table, the change in the selected bond lengths is very small at the states (singlet and triplet) considered. The same holds from BP-DMB and TBP-DMTB. However, as earlier noted, there is a significant difference in bond length moving from benzophenone (i.e., C=O) to thiobenzophenone (i.e., C=S) compounds. The observed bond length for C=O and C=S agrees with the study of Kupka et al.9. For most of the bonds examined, especially those closer to the carbonyl and the thiocarbonyl, it follows that there is a marked change in length, the fact easily explained from the chemistry of S and O. However, it is observed that the farther away from the carbonyl and the thiocarbonyl the bond length, the less pronounced their effect (i.e., the change is minimised) and it is eventually observed that at the para position designated by C9-C12, there is nearly no change in bond length from BP to TBP indicating a neutralisation of the effect the C=S has on the structures. Worthy of note also is the fact that the presence of methoxy group (-OCH3) is not immediately felt on their bond length on moving from BP-DMB and TBP-DMTB.
A similar observation was made for the bond angles of the studied compounds where equal angles were made with the two phenyl rings. Generally, the bond where S is involved is higher than those involving O. As earlier observed, there is no consistent trend in the effect the methoxy (-OCH3) group has on bond angles of the compounds.
As expected, there appears to be no change in lengths and angles of the two phenyl groups with respect to the carbonyl and the thiocarbonyl group suggesting that though not symmetric, the orientation of the two phenyl groups towards the carbonyl groups seems same.
Electronic properties of studied systems
Highest Occupied Molecular Orbital (HOMO), Lowest Unoccupied Molecular Orbital (LUMO), Electron Affinity (EA) and Ionisation Potential (IP): In closed-shell Hartree-Fock theory, the first ionisation energy of a molecular system is equal to the negative of the orbital energy of the HOMO10. The theorem is exact in the context of restricted Hartree-Fock theory if it is assumed that the orbitals of the ion are identical to those of the neutral molecule. The IP calculated this way are in quantitative agreement with experiment. A similar theorem exists in DFT for relating the exact first vertical Ionisation Potential (IP) and Electron Affinity (EA) to the HOMO and LUMO energies. Table 3 and 4 shows the results for HOMO, LUMO, IP and EA calculated with DFT B3LYP/6-31+G (d) and B3LYP/6-31G (d, p) basis sets.
From Table 3, the effect of the addition of substituent (OCH3) and change in functional group (i.e., carbonyl C=O to thiocarbonyl C=S) is immediately seen from their energy gaps. It is known that substituent groups with unshared electron pairs on the atom adjacent to the benzene ring are stronger activating groups and any factor that decreases the energy of the transition state relative to that of the reactants lowers the free energy and increases the rate of the reaction11.
| Table 3: | Singlet-triplet electronic properties of BP, TBP, DMB and DMTB at B3LYP/6-31+G* |
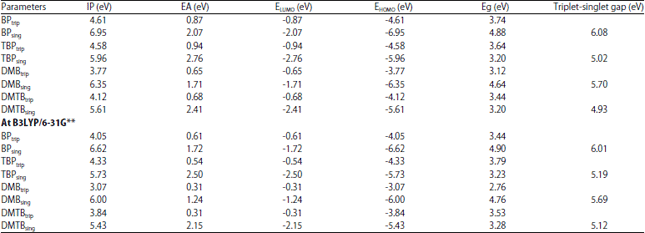 | |
| *B3LYP/6-31+G (d) and **B3LYP/6-31G (d, p) | |
| Table 4: | UV absorption properties of BP of singlet (triplet) calculated at B3LYP TDDFT/6-31+G (d) |
 | |
The presence of the methoxy substituent (OCH3) which is an electron donating group releases electron into the system, stabilises the intermediate state and activate the system thereby lowering the energy gap (singlet-singlet and singlet-triplet) observed in DMB relative to BP. From the understanding of the geometry of sulfur containing compounds, carbon-sulfur double bond, C=S is weaker than C=O due to the size of S preventing proper overlap and interaction of the orbitals unlike when S is single bonded. The triplet LUMO-HOMO smaller band gap observed in DMTB compared with DMB manifests a poor overlap of C=S and an ease in the breaking of the bond thereby enhancing a relatively higher degree of charge transfer. A similar explanation follows for what was observed in BP and TBP differing only in the constitution of their carbonyl group. The TBP as expected has the least singlet-triplet gap of the studied compounds followed by DMTB, establishing a high likelihood of the thiocarbonyl, C=S, taking precedence over methoxy group in contributing to the lowering of the energy gaps as observed. The HOMO and LUMO are the main orbitals taking part in chemical reaction. The HOMO is the outermost (highest energy) orbital containing electrons that could act as an electron donor. The LUMO is the innermost (lowest energy) orbital that has room to accept electrons and can act as an electron acceptor, this is also in agreement with the study of Adejoro et al.12. According to molecular orbital theory, the formation of a transition state is due to an interaction between HOMO and LUMO (Fig. 2). High value of HOMO energy is likely to indicate a high tendency of electron donation to appropriate acceptor molecule of low empty molecular orbital energy giving the order of electron donation from 6-31+G (d) basis set as DMBtrip>DMTBtrip>TBPtrip>BPtrip>DMTBsing>TBPsing> DMBsing>BPsing suggesting among other things the order in which the triplet states (intermediate states) of the compounds will donate electron to attain stability. A lower value of LUMO energy indicates a higher probability to accept electrons13; giving the order of electron acceptance as TBPsing DMTBsing>BPsing>DMBsing>TBPsing>BPtrip>DMTBtrip>DMBtrip. The difference in energy between the two frontier orbitals (HOMO/LUMO) can be used to predict the strength and stability of complexes formed by the compounds as well as the colours they produce in solution hence it follows the conclusion drawn that in general, DMTB with energy gap next to TBP reacts with the entire series of group IVB organometallic radicals with a much better efficiency than does DMB in solution14.
The two basis sets employed in carrying out the calculations gave a close result except for a slight deviation observed in the triplet state of the HOMO of TBP and BP where there is a slight inconsistence.
The IP and EA of an atom or a molecule are the energy needed to remove or gain an electron and therefore can be referred to as the ability of the molecule to donate and accept electrons15, which agrees with the observations seen from their energy gaps as explained above.
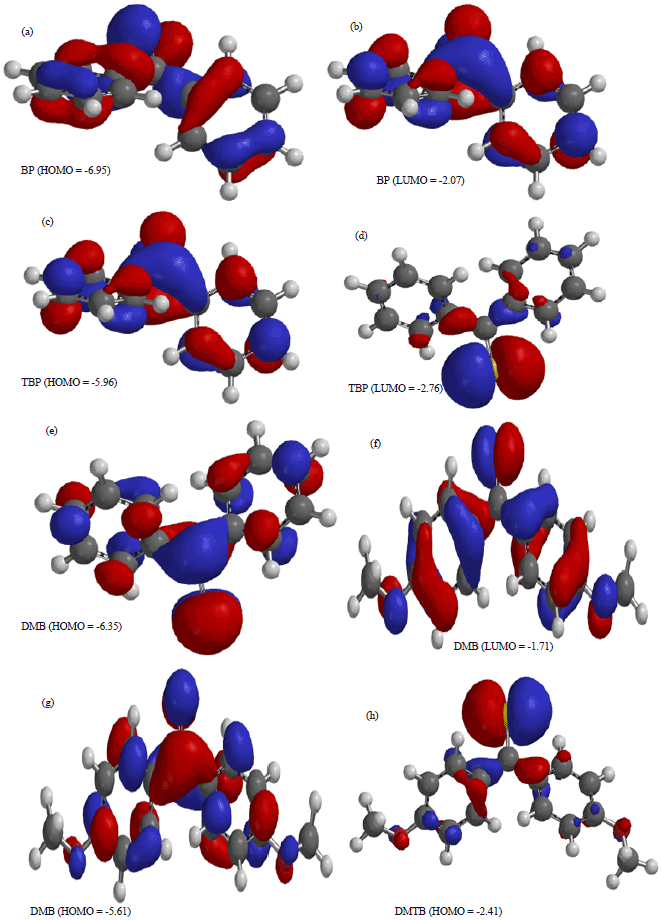 | |
| Fig. 2(a-h): | Frontier molecular orbitals of BP, TBP, DMB and DMTB |
The singlet state of BP with the highest IP (6.62 eV) indicates the least readiness for electron removal whereas DMTB (singlet) with the lowest IP (3.84 eV) suggests the highest ease of electron removal from its molecule thereby a higher intramolecular charge transfer is expected in its molecule.
Electronic transitions: The total energy, energy gap and dipole moment have influence on the stability of molecule. Optimisation was performed in order to investigate the energetic behaviour and dipole moment of title compounds. The UV spectrum of BP was taken both in n-hexane and ethanol alongside calculations done on all the studied compounds using B3LYP TDDFT/6-31+G (d). In the benzophenone, both phenyls can interact with the C=O group through σ (inductive effect) bond and π (mesomeric effect) bond. The overlapping between the π bonds of the rings and of the C=O form a molecular orbital that comprises the entire molecule. Due to this π-electronic delocalisation, the C=O group loses part of its individual character and partially integrates with phenyls leading to system stabilisation and transference of the electronic deficiency from the atom of Ccarbonylic to the molecule1. Therefore, band due to pure C=O (n→π*) absorption whose peak is expected around 290 nm could not be observed at 290 nm due to the interaction with the phenyl ring. The UV spectral of benzophenone exhibits two bands in the two solvents. The bands exhibited in n-hexane are I: 205.3 and II: 247.6 while, the bands exhibited in ethanol are I: 197.7 and II: 252.7. The shifts of the bands are Δλ = -7.6 (i.e., non-polar to polar) and Δλ = +4.7. The Δλ for band I is due to π→π* transition while Δλ = -7.6 corresponds to n→π*. The blue shift of band I is due to H-bonds between solute and solvent (Fig. S1, S2). According to Frank-Condon principle, the maximum absorption peak (λmax) in UV-visible spectrum corresponds to vertical excitation. The HOMO is the orbital that acts primarily as an electron donor and LUMO is the orbital that acts largely as electron acceptor. The energy gap of HOMO-LUMO explains the eventual charge transfer interaction within the molecule6 which influences the photochemical activity of the molecules.
It is worthy of note that the triplet states of the studied compounds do not absorb in the UV-Vis region of the electromagnetic (EM) radiation as seen from Table 4-7 which could be an hint to the nature of their ground state as the compounds are expected to absorb in the UV-Vis region of the EM radiation because of their relatively high conjugation. The fact that the triplet states are not absorbing from UV-V is region from the calculations performed suggests that there is a disruption of conjugation which is due most likely to the fact that they are no more at the conjugated state able to so absorb at the region-the effect that would be expected if electron is abstracted from the molecule of the compounds (i.e., in radicals) thereby distorting conjugation.
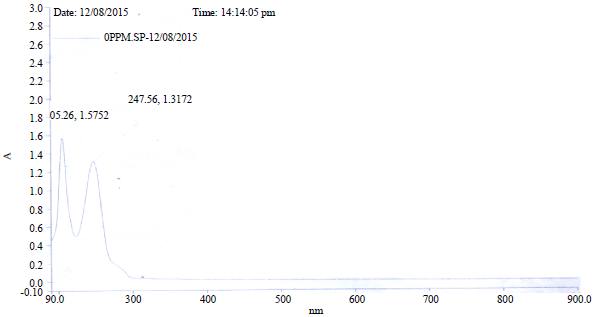 | |
| Fig. S1: | Experimental UV-Vis spectrum of BP in n-hexane |
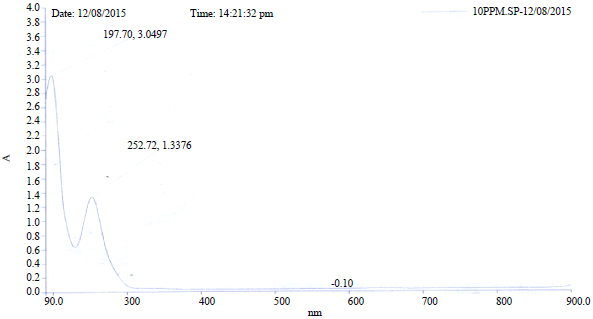 | |
| Fig. S2: | Experimental UV-Vis spectrum of BP in ethanol |
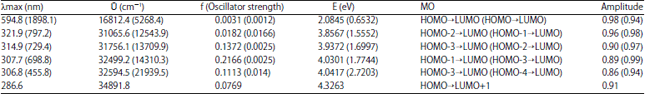
| Table 5: | UV absorption properties of TBP singlet/(triplet), calculated at B3LYP TDDFT/6-31+G (d) TBP |
 | |
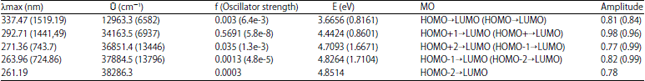
| Table 6: | UV absorption properties of DMB singlet/(triplet), calculated at B3LYP TDDFT/6-31+G (d) |
 | |
| Table 7: | UV absorption properties of DMTB singlet/(triplet) calculated at B3LYP TDDFT/6-31+G (d) |
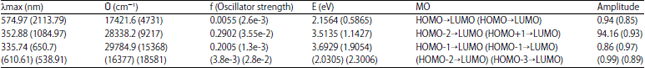 | |
Also, it is observed that transitions from calculations are more than that observed in BP which may likely be the case with TBP, DMB and DMTB when experimented. This is due in part to the solvent used and other factors which may not be immediately noted but summed up in the fact that not all the calculated transitions are allowed. Some transitions are quantum mechanically forbidden while others are overlap forbidden; both of which are functions of symmetry and multiplicity of the ground and excited state of the orbital concerned16. The presence of thio group in BP leads to an increase in absorption wavelength of 594.8 nm, which deceases at transitions. The absorption wavelength of 307.7 nm which corresponds to an oscillatory strength of 0.2 is an allowed transition with the promotion of electron HOMO-1→LUMO compared to BP may also be due to the nature of symmetry of the system (Fig. 3). A quick inspection of the above gives a hint of a high probability of spectra overlap.
It is reported that the singlet transition of the parent DMTB molecule lie in the visible region while electronic transition in DMB molecules are mainly in the UV region of EM radiation14, the fact immediately evident from the computed absorption properties tabulated above.
 | |
| Fig. 3(a-d): | Calculated UV spectra of (a) BP, (b) TBP, (c) DMB and (d) DMTB |
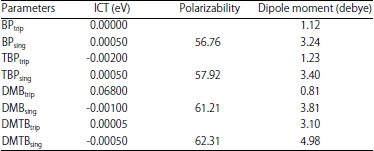
Dipole moments and polarizabilities: Dipole moment reflects the molecular charge distribution in a molecule. Dipole moments and polarizabilities are the molecular properties that characterises the response of a system in an applied electric field. They determine not only the strength of molecular interactions (reactivity), the cross section of different scattering and collision processes but also the electro-optic (non-linear optical) properties of a system17-19. These properties for the studied compounds are presented in Table 8. It is observed that the dipole moments calculated has a value corresponding to polarizabilities of the singlet states. From the results obtained, it is expected that the singlet state of DMTB will be most reactive of all the singlet states-an observation which is in consonance with what was observed from their energy gap.
IR vibrational assignment: The BP has 22 atoms with symmetry of C2V, hence, it has 60 normal modes of vibration. In order to obtain the spectroscopic assignment of BP, the wave number calculation analysis is performed using DFT (B3LYP). Calculations were made for gaseous phase whereas the experiments were performed for solid phase. Therefore, there is some disagreement between the calculated and observed vibrational wave numbers.
The calculated harmonic force constants and wave numbers are usually higher than the corresponding experimental quantities because of the combination of electron correlation effects and basis set deficiencies.
| Table 8: | Intramolecular charge transfer, dipole moments and polarizabilities of the studied compounds calculated at DFT B3LYP/6-31+G (d) |
 | |
The observed slight disagreement between theory and experiment could be a consequence of the anharmonicity and the general tendency of the quantum mechanical methods to overestimate the force constants at the exact equilibrium geometry. Some of the vibrations observed are C-H vibrations, Methyl group vibrations, C-C vibrations and C=O vibrations.
The studied compounds contain methoxy (OCH3) groups. In such molecules, electronic charge is back donated from the lone pair of electrons on oxygen atom to the σ* orbital of C-H bonds causing weakening of the C-H bonds. This is followed by an increase in C-H bond distance and the decrease in C-H force constant. This can result in enhancement of IR band intensities of the C-Hstretching modes. The CH3 group has several modes associated with it such as symmetric and asymmetric stretching, bends, rocks and torsions. Both the asymmetric and symmetric deformations of these methyl groups are calculated in the range 1475-1432 cm–1. The rocking mode of vibration of CH3 group usually appears in the region 1070-1010 cm–1 Raj et al.20. The rocking modes are calculated below 1192 cm–1.
The experimental C-Hstretching was observed at 3289 and 3226 cm–1 characteristic of Csp2-H hybridisation (Fig. S3, S4). The theoretical vibrational frequency was calculated as 3218 cm–1 (Fig. 4).
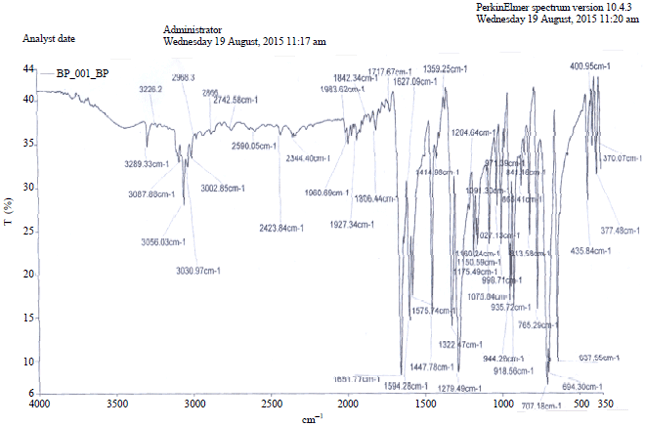 | |
| Fig. S3: | Experimental FT-IR spectrum of BP using KBr disc |
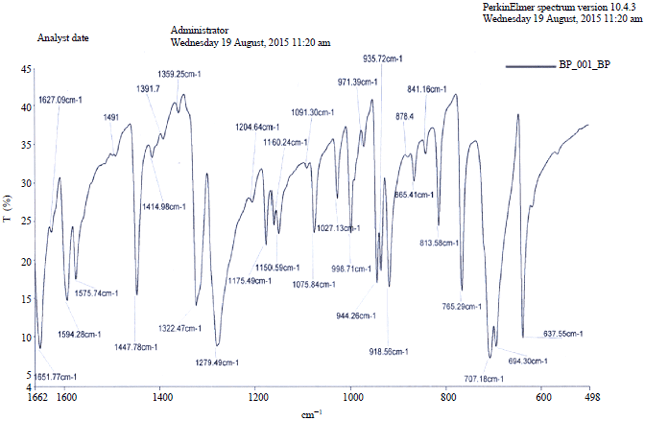 | |
| Fig. S4: | Expanded FT-IR spectrum of BP |
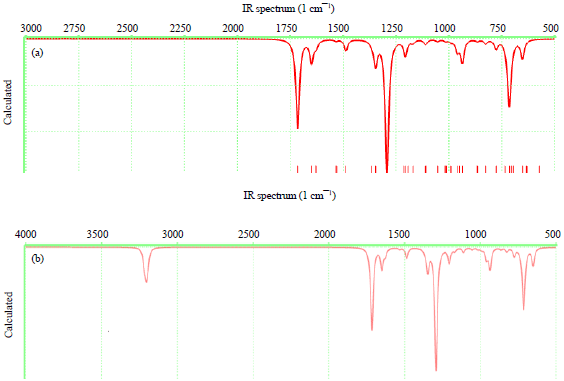 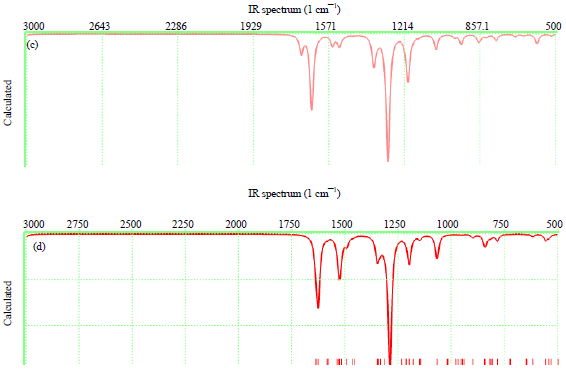 | |
| Fig. 4(a-d): | Calculated IR spectra of (a) BP, (b) TBP, (c) DMB and (d) DMTB |
The peak is short but sharp, a characteristic region for ready identification of C-Hstretching vibration this is in concordance with that reported by Adejoro et al.21.
Most aromatic compounds have nearly four IR peaks in the region 3080-3010 cm–1 due to ring C-Hstretching bonds. The corresponding peaks observed are 3088 cm–1 (w), 3056 cm1–(m), 3030 cm–1 and 3003 cm–1 (w). There was no such corresponding peaks in calculation but peaks between 3200 and 3100 cm–1 (4 peaks) can be said to agree with the above bearing in mind what has been earlier noted. The ring C-Hstretching appears to be very weak. This is due to steric interaction that induces effective conjugation and charge carrier localization resulting in twisted phenyl ring. In this region, the bands are not affected appreciably by the nature of substituent. The C-H in-plane deformation is calculated at 1153 cm–1 with high intensity. The C-H out-of-plane deformation is calculated at 892 and 862 cm–1. The phenyl ring asymmetric, symmetric deformation is calculated at the coupled vibrations below a range of 650 cm–1. There are two methoxy groups attached to the phenyl rings each of DMB and DMTB. The C-O in-plane bending is calculated at 607 and 286 cm–1 in the highly mixed modes. The C-O out-of-plane bending vibrations are calculated at 708 and 505 cm–1 C-H in-plane bending vibrations have strong and medium intensity bands in the region 1450-1000 cm–1.
For C-C vibrations, the ring stretching vibrations are very much important in the spectrum of BP and its derivatives. The aromatic ring C-C stretching vibrations occur in the region 1650-1430 cm–1 which agrees with the experimental observations as peaks strong to medium appearing as four bands of frequency 1652, 1627, 1594 and 1576 cm–1 and also in conformity with the study of Adejoro et al.21.
The C=O bond formed by π-π interaction between C and O. Internal hydrogen bonding reduces the frequencies of the C=O stretching absorption to a greater degree than does intermolecular hydrogen bonding. Because of the different electronegativities of C and O, the bonding is not equally distributed between the two atoms.
CONCLUSION
The UV-Vis and FT-IR spectra of BP have been recorded and the vibrational assignments have been carried out. Electronic absorption bands, energy gaps and IR spectra, the intermolecular charge transfer between HOMO and LUMO energies, frontier energy gap of BP, DMB and DMTB were also computed. The difference between the experimental and computed wavelengths and wavenumbers is small and falls within the range reported in literatures and experimental observations. From this study, it is established that DMB and DMTB have more than one conformers while BP showed only one conformer, there is likelihood that DMTB with the lowest energy gap will exhibit highest charge transfer of the three though this is subject to further studies. The singlet state of DMTB with the lowest IP suggests the highest ease of electron release, while BP with the highest IP indicates the least readiness for electron release.
The present experimental and quantum mechanical study provides important role in the understanding of dynamics and nature of ground electronic states of these molecules. All the compounds studied are potential non-linear optical (NLO) materials which can be applied in photovoltaic cells. Although, the computation gives a clue of what is to be expected, more experimental work needs to be done particularly on DMTB which promises to be a good optical material.
REFERENCES
- Castro, G.T., S.E. Blanco and O.S. Giordano, 2000. UV spectral properties of benzophenone. Influence of solvents and substituents. Molecules, 5: 424-425.
CrossRefDirect Link - Fisera, L., R. Huisgen, I. Kalwinsch, E. Langhals and X. Li et al., 1996. New thione chemistry. Pure Applied Chem., 68: 789-798.
CrossRefDirect Link - Sustmann, R., W. Sicking and R. Huisgen, 2003. A computational study of the cycloaddition of thiobenzophenone S-methylide to thiobenzophenone. J. Am. Chem. Soc., 125: 14425-14434.
CrossRefPubMedDirect Link - Ibeji, C.U., I.A. Adejoro and B.B. Adeleke, 2015. A benchmark study on the properties of unsubstituted and some substituted polypyrroles. J. Phys. Chem. Biophys., Vol. 5.
CrossRefDirect Link - Shao, Y., L.F. Molnar, Y. Jung, J. Kussmann and C. Ochsenfeld et al., 2006. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys., 8: 3172-3191.
CrossRefPubMedDirect Link - Kupka, T., R. Wrzalik, G. Pasterna and K. Pasterny, 2002. Theoretical DFT and experimental Raman and NMR studies on thiophene, 3-methylthiophene and selenophene. J. Mol. Struct., 616: 17-32.
CrossRefDirect Link - Chow, M.S., L.V. Liu and E.I. Solomon, 2008. Further insights into the mechanism of the reaction of activated bleomycin with DNA. Proc. Natl. Acad. Sci., 105: 13241-13245.
CrossRefDirect Link - Adejoro, I.A., D.C. Akintayo and C.U. Ibeji, 2016. The efficiency of chloroquine as corrosion inhibitor for aluminium in 1M HCl solution: Experimental and DFT study. Jordan J. Chem., 11: 38-49.
Direct Link - Adeleke, B.B., K.S. Chen and J.K.S. Wan, 1981. A comparative esr study of the reactions of group ivb organometallic radicals with 4,4′-dimethoxybenzophenone, 4,4′-dimethoxythiobenzophenone and 3-ethyl-2-thioxo-4-oxazolidione. J. Organometallic Chem., 208: 317-326.
CrossRefDirect Link - Ullah, H., A.H. Ali Shah, K. Ayub and S. Bilal, 2013. Density functional theory study of poly (o-phenylenediamine) oligomers. J. Phys. Chem. C, 117: 4069-4078.
CrossRefDirect Link - Adeoye, M.D., N.O. Obi-Egbedi and I. Iweibo, 2012. Solvent effect and photo-physical properties of 2,3-diphenylcyclopropenone. Arabian J. Chem.
CrossRefDirect Link - Anbarasan, P.M., P.S. Kumar, M. Geetha, R. Govindan, S. Manimegalai and K. Velmurugan, 2010. Geometries, electronic structures and electronic absorption spectra of silicon dichloride substituted phthalocyanine for dye sensitized solar cells. Recent Res. Sci. Technol., 2: 8-16.
Direct Link - Sundaraganesan, N., J. Karpagam, S. Sebastian and J.P. Cornard, 2009. The spectroscopic (FTIR, FT-IR gas phase and FT-Raman), first order hyperpolarizabilities, NMR analysis of 2,4-dichloroaniline by ab initio HF and density functional methods. Spectrochim. Acta. Part A Mol. Biomol. Spectrosc., 73: 11-19.
CrossRefPubMedDirect Link - Kosar, B., C. Albayrak, C.C. Ersanli, M. Odabasoglu and O. Buyukgungor, 2012. Molecular structure, spectroscopic investigations, second-order nonlinear optical properties and intramolecular proton transfer of (E)-5-(diethylamino)-2-[(4-propylphenylimino)methyl]phenol: A combined experimental and theoretical study. Spectrochim. Acta Part A: Mol. Biomol. Spectrosc., 93: 1-9.
CrossRefDirect Link - Raj, A., K. Raju, H.T. Varghese, C.M. Granadeiro, H.I.S. Nogueira and C.Y. Panicker, 2009. IR, Raman and SERS spectra of 2-(methoxycarbonylmethylsulfanyl)-3,5-dinitrobenzene carboxylic acid. J. Braz. Chem. Soc., 20: 549-559.
CrossRefDirect Link - Adejoro, I.A., E. Akintemi, O.O. Adeboye and C. Ibeji, 2014. Quantum mechanical studies of the structure-activity relationship and electronic vibration of some dietary flavonoids. Asian J. Applied Sci., 7: 117-128.
CrossRefDirect Link