
Topik Hidayat
Department of Biological Sciences, Faculty Bioscience and Bioengineering, Universiti Teknologi Malaysia, Johor Bahru, Malaysia
Mohd. Aszuan Abdul Samat
Department of Biological Sciences, Faculty Bioscience and Bioengineering, Universiti Teknologi Malaysia, Johor Bahru, Malaysia
Muhamad Atiq bin Elias
Department of Biological Sciences, Faculty Bioscience and Bioengineering, Universiti Teknologi Malaysia, Johor Bahru, Malaysia
Tony Hadibarata
Institute of Environmental and Water Research Management, Universiti Teknologi Malaysia, Malaysia
Journal of Applied Sciences
Year: 2012 | Volume: 12 | Issue: 4 | Page No.: 354-361







ABSTRACT
Index for water quality is solely based upon the conditions of physical and chemical which provide quantitative data on the presence and level of aquatic pollution. However, the parameters for the assessment do not represent the environmental stress and ecological health of the river which affect the microbial diversity. In this study, we introduced a new approach of assessing river water from metagenomic view by constructing phenetic tree which show relation between microbial community and the level of pollution. Four river waters, of which one river is categorised as unpolluted water, located around Johor Bahru (Malaysia) were examined and compared. Genomic DNA of these uncultured community of microorganisms was directly extracted and 16S rRNA gene was amplified by PCR using a set of primer pairs to generate cloned libraries. In total 24 isolates which are consisted of 18 isolates from polluted rivers and six from unpolluted one, were sequenced. Along these sequences, six of 16S rRNA sequences of colliform bacteria obtained from genebank were placed in Operational Taxonomic Units (OTUs). Phenetic analysis revealed that the river water used in this study were classified into two groups, representing the polluted and unpolluted. Since the tree can clearly distinct unpolluted river water from that of polluted one, we tried to develop a putative sequences motif for both condition. In practical purpose, the sequences motif can be used to screen river water quality in certain river water environment.
PDF Abstract XML References Citation
Received: December 06, 2011;
Accepted: January 28, 2012;
Published: March 21, 2012
How to cite this article
Topik Hidayat, Mohd. Aszuan Abdul Samat, Muhamad Atiq bin Elias and Tony Hadibarata, 2012. Metagenomic Analysis of 16S rRNA Sequences from Selected Rivers in Johor Malaysia. Journal of Applied Sciences, 12: 354-361.
DOI: 10.3923/jas.2012.354.361
URL: https://scialert.net/abstract/?doi=jas.2012.354.361
DOI: 10.3923/jas.2012.354.361
URL: https://scialert.net/abstract/?doi=jas.2012.354.361
INTRODUCTION
The ecological distress due to the contaminated freshwater sources by human activity leads to the importance of monitoring the freshwater status. Water quality assessments are usually carried out by doing the physico-chemical analysis, represented by Water Quality Index (WQI) (Agarwal and Rajwar, 2010; Nasab et al., 2010) and sometimes include the microbiological study (Trivedy and Goel, 1984). However, this physical and chemical measurement does not reflect the extent of environmental stress reaching the living organisms or the subsequent effects of the stress towards the organisms in that environment (Kerkhof, 2009; Maznah and Omar, 2010; Oparaku et al., 2011). The use of metagenomics approach has offered in comprehensive environmental assessment of microbial community in water environment (Cottrell et al., 2005; Marshall et al., 2008), reflecting of microbial diversity under environmental stress.
Metagenomics, or also known as environmental genomics, is study of collective genomes of microorganisms. By using metagenomics, we are able to perform genomic analysis of microorganisms by direct extraction and cloning of DNA from an assemblage of microorganisms, collected from various types of environment such as soil, river, feces and marine (Handelsman, 2004; Bhuiyan et al., 2011). This is therefore unnecessary to collect and culture the microorganisms and allows us to avoid the bias imposed by culturing and led to the discovery of vast new lineages of microbial life (Daniel, 2004; DeLong et al., 2006). It is now widely accepted that the application of standard microbiological methods for the recovery of microorganisms from the environment has had limited success in providing access to the true extent of microbial biodiversity. It follows that much of the extant microbial genetic diversity remains unexploited, an issue of considerable relevance to a wider understanding of microbial communities. The recent development of molecular biology designed to access this wealth of genetic information through environmental nucleic acid extraction has provided a means of avoiding the limitations of culture-dependent genetic exploitation (Cowan et al., 2005).
In this study, we conducted metagenomic analysis of 16S rRNA gene, in particular, to evaluate utility of the sequences information in assessing river water quality. With its high level of conservative sequences, the 16S rRNA gene has been the main source of genetic material used to study comparative genomic analysis since long (Weisburg et al., 1991; Devereux and Mundfrom, 1994; Farrelly et al., 1995). Thus, we compared metagenomic sequences from different site of river water (polluted and unpolluted) and subjected them to phenetic analysis. Phenetic relationships groups the organisms together based on overall similarity of their phenotypic (nucleotides) characteristic which are observable. Molecular approach to study water quality can widen our perspective in assessing pollution level of the river, for instance, by reconstructing a sequences motif and oligonucleotide primers/probes which have the potential utility to assess rapidly the environmental health condition and status in the future application.
MATERIALS AND METHODS
Four rivers, namely, Kota Tinggi (CN), Sayong (SY), Skudai (SK) and Kempas (KM) were selected. This selection was made based upon the information issued by local government (Department of Environment/DOE and Department of Irrigation and Drainage/DOIAD, Johor, Malaysia). According to this report, CN is subjected to unpolluted, whereas the rest of three rivers are polluted. A water sample was collected from a depth of 50 cm in July 2011.
Genomic DNA from the selected rivers was extracted by using the CTAB method developed by Marshall et al. (2008) with a little modification. About 5 L water samples were filtered by using aspirator pump. The samples were filtered through 0.2 mm microfibre filter paper that was placed in 2 mL CTAB extraction buffer before incubate them in water bath for 70°C for at least 1 h. Then, the filter papers were removed from the buffer. Each 1 mL of the buffer was transferred to the 2 mL tube. Then, 1 mL 24:1 (v/v) Chloroform:Isoamyl alcohol was added, the tube mix by inverted several times before centrifuge for 10 min at 13,200 rpm. The supernatant was transferred into the new 1.5 mL tube, 0.7 volumes of 100% cold 2-Propanol were added, the tube mixed by inverting several times. The tube was subsequently centrifuged for 30 min at 13,200 rpm at room temperature. The supernatant were decanted, whereas the pellet was kept and leave air-dried by opened the tube about 10-15 min or until the pellet dried. The pellet was subsequently rehydrated in 25 μL TE buffer pH 8.0 and stored in -20°C for long term usage. In some cases, purification was performed with a HiYield™ Gel/PCR Mini Kit following the manufacturer’s instructions.
The amplification of the 16S rRNA gene was carried out using the primer pairs pAf and pHr (Edwards et al., 1989; Bruce et al., 1992), forward and reverse, respectively. Each reaction included 5 μL 10x Standard Taq reaction buffer, 1 μL of each 10 μM forward and reverse primers, 1 μL 10 μM dNTPs, 39.75 μL sterile deionized water, 0.25 μL Taq DNA polymerase and 2 μL template genomic DNA. The PCR profile consisted of an initial 2 min premelt at 94°C and 30 cycles of 1 min at 94°C (denaturation), 1 min at 45°C (annealing) and 2 min at 72°C (extension), followed by a final 2 min extension at 72°C. PCR products were visualized by 0.8% of agarose gel electrophoresis.
Ligation was set up by mixing 5 μL of PCR product with 5 μL of 2 X rapid ligation buffer, 1 μL of T4 DNA ligase and 1 μL pGEM-T vector. The reaction were mixed by pipetting and incubated for overnight at 4°C in refrigerator. Before starting the transformation procedure, the incubator shaker was preheated at 37°C and water bath at 42°C. Ice also was prepared. The ligation mixtures were removed from the refrigerator and equilibrated to room temperature for 1 min and centrifuged briefly. Then 6 μL of each ligation mixture were added to 1.5 mL tube which has been pre cooled on ice. Then the DH5α Competent cells were removed from freezer and placed in a 50% ice/50% distilled water bath for 5 min. Then 150 μL of competent cells were added to the tubes on ice. The mixture then leaved on ice for 20 min. Then, heatshocked the cells for 45 sec at 42°C and returned to ice for 2 min. 300 μL of LB media were added to the tube and mixed by flicking gently. After that, the tube was closed completely [airtight] and put in incubator shaker at 37°C [optimum temperature for bacteria growing] for 90 min. During the incubation process, the LAIX plates were prepared and put in 37°C incubator oven to dry out for 30 min. After the plates were dried and transformation culture incubated, the plates then were taken to the laminar flow hood. Then, 150 μL of the transformation cultures were transferred onto the plates and spread by using hockeystick. The plates were sealed with parafilm and placed in 37°C oven for 20 h. After the LAIX plates incubation of 20 h, the colonies were screened to select white colonies instead of blue (possible positive colonies that have an insert of gene interest).
About 50% from total white colonies were selected and labeled. By using sterile pipette tips, each single white colony was picked and put a drop on the labeled grid plate (for PCR library construction) and then dipped in the 5 mL LB medium (for plasmid extraction). The grid plates then were incubated for 20 h and 37°C before stored at 4°C. Plasmid extraction was conducted using QIAmp Spin Miniprep following the manufacturer’s instructions.
The restriction enzyme digestion was conducted using EcoRI to reconfirm whether the plasmid has the insert desired or not. Digestion mixture were added in 1.5 mL tube and mixed together by pipetting before adding EcoRI. The mixtures were gently mixed by pipetting and incubated for 4 h at 37°C. As many as 24 isolates/clones which consist of 18 isolates/clones from polluted river (SY, SK dan KM) and six from unpolluted one (CN) were sent to 1stBase for sequencing.
DNA sequences obtained were edited and assembled using CodonCode Aligner (http://www.codoncode.com/aligner/). Multiple alignments were conducted using ClustalX (www.clustal.org/). In total 30 OTUs (of which 24 obtained from this study plus six sequences retrieved from Genebank) was subjected to phenetic analysis. The 30 aligned sequences were used to construct the phenetic tree with MEGA version 4 (www.megasoftware.net). After the construction of the phenetic tree, a putative DNA sequences motifs for both unpolluted and polluted group was developed from the consensus sequences which were identified by GENEIOUS (www.geneous.com/).
RESULTS AND DISCUSSION
The aligned 16S RNA gene comprised 1,544 characters. Phenetic analysis using UPGMA (Unweighted Pair Group Method with Arithmetic Mean) clustering method resulted in a single tree (Phenogram) as depicted in Fig. 1. The tree demonstrated that the OTUs comprises two groups which represent two conditions of river, unpolluted and polluted group. The polluted group is housed by mixtures among three examined rivers (SY, SK and KM) and the existence of the coliform bacteria (Klebsiella spp., Hafnia spp., Serratia spp., Enterobacter spp., Citrobacter spp. and Escherichia coli) supported the grouping.
The term “sequences motif” here refers to DNA sequences of the 16S rRNA gene present in the sample that is unique and can be used to capture and record environmental condition under ecological surrounding in particular time. This term can be compared with other term such as “metagenomic profile”.
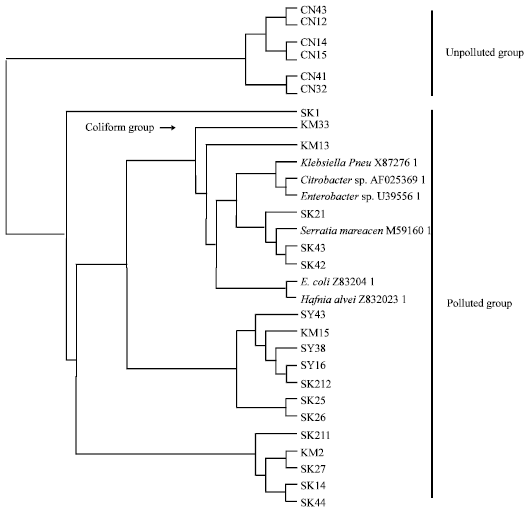 | |
| Fig. 1: | Phenetic tree derived from 24 isolates which were consisted of 18 from polluted rivers (SY, SK and KM) and six from unpolluted one (CN). Six coliform bacteria were embedded in polluted group |
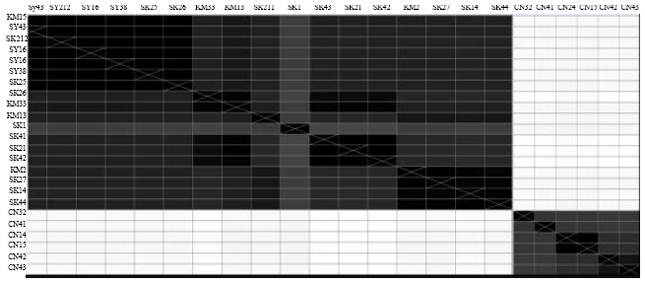 | |
| Fig. 2: | The heat map of 24 isolates generated by GENEIOUS software. Distances are represented by shades of gray. A lighter gray means further apart while darker means closer together. The longest distance in the tree is white and the shortest distance is black (may not be zero) |
As the tree can clearly distinguish unpolluted river water from that of polluted one, a putative sequence motif was made, as can be seen in Fig. 3 and 4.
This study focused on determining the overall similarity of microbial communities among different level of river water pollution. They were compared with each other and discerned a significance differences at molecular level. The phenetic tree (Fig. 1) separated distinctly unpolluted bacterial groups from those polluted group. This grouping is consistent with what has been reported by local government (DOE and DOIAD) that three rivers, notably, SY, SK and KM are polluted, whereas CN is unpolluted.
To confirm this, we included the 16S rRNA sequences of the coliform bacteria from GeneBank in the phenetic tree reconstruction. As result, all the coliform species used were housed in polluted group. The reason why we included the gene sequence from coliforms is because the coliforms always refer as indicator organisms when assessing water quality (Kovacs, 1992; Ghandhari and Alavi Moghaddam, 2011; Durai and Rajasimman, 2011). The coliform bacteria have been served as bioindicator for potential presence of disease-causing bacteria in water. This group of bacteria is common in soil and surface water as it also abundance in waste from humans and animals. The presence of coliform indicates that the water were polluted either cause of water discharge from farm, drainage or industrial waste.
Phenetic relationship shows that the organisms that share the same characteristics will group together. In this case, perhaps, the organisms have the same characteristics at molecular level which will help them to adapt at certain environment (Herrera and Cockell, 2007).
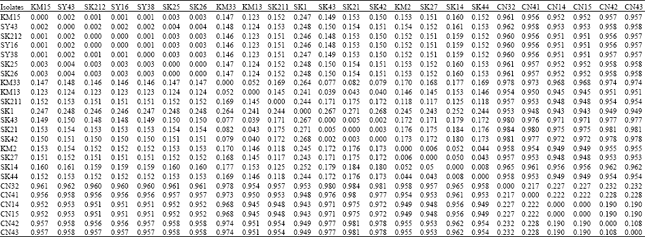
The genetic similarity between OTUs was also determined by converting the results to the heat map (Fig. 2) and distance matrix (Table 1). The patristic distances were calculated by summing together the lengths of the branch between each pair of OTUs. The value of the distance matrix based on fixation index which the value range is in between 0 to 1. The value 0 indicates that the isolates were genetically identical while the value 1 indicates that the isolates were completely separated or different. Based on Fig. 3, it is clearly showed that the samples representative of unpolluted and polluted group were significantly different with each other due to the range value of unpolluted to polluted group were more than 0.9. The isolates which have the range within 0.1±0.05 can be considered in the same group (Kwoon, 2000). This result suggested that each of the microbial community which represent unpolluted and polluted group having different set genetic data of certain environment condition, either the microbial community have particular site of nucleotide or amino acid which enable them to survive in polluted condition or the deletion/insertion of certain DNA location may absence/presence due to the environmental stress.
| Table 1: | The patristic distance matrix of 24 isolates |
 | |
 | |
| Fig. 3: | Putative sequence motif for unpolluted river water sample derived from consensus sequences |
Pollution caused by pollutants will result in environmental stress. This situation will inevitably affect the genome structure of microbial community. Miller (1993) and Al-Sheikh and Fathi (2010) reported that environmental stress will increase the genetic alteration by mutation. Thus, there is potential and chances of the mutation within a specific condition (e.g., polluted) environment such as aquatic ecosystem (Praveena et al., 2011; Ahmed, 2012).
Exchanges genetic material between natural microbial communities appears to be common phenomenon (Ford, 1994; Ahmed, 2012). The exchanging will mediated by several microbial processes either by conjugation, transformation and transduction. Microbial diversity adaptation towards environmental stress involve either by selective species enrichment, induction/derepression of enzymes or by genetic changes. Bacteria have the ability to rapidly tolerate and adapt toxic substances in their environment (Ford, 1994; Amann et al., 1995; Ahmed, 2012).
The data and information obtained from the alignment can be used to develop a putative sequence motif for polluted and unpolluted group which is derived from consensus sequences (Fig. 4). These putative motifs are unique for each group. With their term “metagenomic profile”, Marshall et al. (2008) suggested that this “profile” can be used to characterize ecosystems with some difficulties in implementing it in the field. The difference between our study and that of Marshall et al. (2008) is that this study are trying to facilitate river quality assessment using molecular screening through the sequences motifs reconstruction.
This samples number were small but present study suggest that even with the small number of samples, the molecular assessment of aquatic ecosystem can be conducted to characterize the ecological health situation. This is because our phenetic tree obviously shows the two main alliances representing the polluted and unpolluted group. Even the coliform groups were embedded in the polluted group.
 | |
| Fig. 4: | Putative sequence motif for polluted river water sample derived from consensus sequences |
This proved that although in small number of samples, the microbial community still capable to shows their varied tolerance ranges, in this case their adaptability towards the pollution level.
Although, the study is considered to be preliminary, our results suggest that the metagenomic analysis of microbial communities have the potential to characterize ecosystem which are differ from each other. Microbial communities can provide useful information about ecological health status which cannot be told by chemical and physiological indicators. Information such as microbial interactions within community and putative sequences motif which are unique for each environment situation can be retrieved. Although there are lot of studies must be conducted about to optimize the methodology, this study shows that this technology can be useful tools for research, environmental agencies and industry. However, further analysis with greater study sites is desirable to establish more robust conclusion.
ACKNOWLEDMENT
This study was funded by Research University Grant from the Universiti Teknologi Malaysia (Vote No. 77553) which is gratefully acknowledged.
REFERENCES
- Agarwal, A.K. and G.S. Rajwar, 2010. Physico-chemical and microbiological study of tehri dam reservoir, Garhwal Himalaya, India. J. Am. Soc., 6: 65-71.
Direct Link - Amann, R.I., W. Ludwig and K.H. Schleifer, 1995. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev., 59: 143-169.
PubMedDirect Link - Bhuiyan, F.A., S. Nagata and K. Ohnishi, 2011. Novel chitinase genes from metagenomic DNA prepared from marine sediments in Southwest Japan. Pak. J. Biol. Sci., 14: 204-211.
CrossRefDirect Link - Bruce, K.D., W.D. Hiorns, J.L. Hobman, A.M. Osborn, P. Strike and D.A. Ritchie, 1992. Amplification of DNA from native populations of soil bacteria by using the polymerase chain reaction. Applied Environ. Microbiol., 58: 3413-3416.
Direct Link - Cottrell, T.M., L.A. Waidner, L. Yu and D.L. Kirchman, 2005. Bacterial diversity of metagenomic and PCR libraries from the Delaware River. Environ. Microbiol., 7: 1883-1895.
PubMed - Cowan, D., Q. Meyer, W. Stafford, S. Muyanga, R. Cameron and P. Wittwer, 2005. Metagenomic gene discovery: Past, present and future. Trends Biotech., 23: 321-329.
PubMed - Daniel, R., 2004. The soil metagenome-a rich resource for the discovery of novel natural products. Curr. Opin. Biotechnol., 15: 199-204.
PubMed - DeLong, E.F., C.M. Preston, T. Mincer, V. Rich and S.J. Hallam et al., 2006. Community genomics among stratified microbial assemblages in the ocean's interior. Science, 311: 496-503.
CrossRefDirect Link - Devereux, R. and G.W. Mundfrom, 1994. A phylogenetic tree of 16S rRNA sequences from sulfate-reducing bacteria in a sandy marine sediment. Applied Environ. Microbiol., 60: 3437-3439.
Direct Link - Edwards, U., T. Rogall, H. Blocker, M. Emde and E.C. Bottger, 1989. Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucl. Acids Res., 17: 7843-7853.
CrossRefPubMedDirect Link - Farrelly, V., F.A. Rainey and E. Stackebrandt, 1995. Effect of genome size and rrn gene copy number on PCR amplification of 16S rRNA genes from a mixture of bacterial species. Applied Environ. Microbiol., 61: 2798-2801.
Direct Link - Ford, T., 1994. Pollutant effects on the microbial ecosystem. Environ. Health Perspect., 102: 45-48.
Direct Link - Handelsman, J., 2004. Metagenomics: Application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev., 68: 669-685.
CrossRefDirect Link - Herrera, A. and C.S. Cockell, 2007. Exploring microbial diversity in volcanic environments: A review of methods in DNA extraction. J. Microbiol. Method, 70: 1-12.
PubMed - Marshall, M.M., R.N. Amos, V.C. Henrich and P.A. Rublee, 2008. Developing SSU rDNA metagenomic profiles of aquatic microbial communities for environmental assessments. Ecol. Indic., 8: 442-453.
CrossRefDirect Link - Maznah, W. and W. Omar, 2010. Perspectives on the use of algae as biological indicators for monitoring and protecting aquatic environments, with special reference to malaysian freshwater ecosystems. Trop. Life Sci. Res., 21: 51-67.
Direct Link - Weisburg, W.G., S.M. Barns, D.A. Pelletier and D.J. Lane, 1991. 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol., 173: 697-703.
CrossRefPubMedDirect Link - Praveena, S.M., S.S. Siraj, A.K. Suleiman and A.Z. Aris, 2011. A brush up on water quality studies of port Dickson, Malaysia. Res. J. Environ. Sci., 5: 841-849.
CrossRefDirect Link