
Muneer Ahmad
College of Computer and Information Sciences, King Faisal University, P.O. Box 55028, Al-Hassa 31982, Saudi Arabia
Azween Abdullah
Department of IT, University Technology PETRONAS, Malaysia
Khalid Buragga
College of Computer and Information Sciences, King Faisal University, P.O. Box 55028, Al-Hassa 31982, Saudi Arabia
Journal of Applied Sciences
Year: 2011 | Volume: 11 | Issue: 5 | Page No.: 806-814







ABSTRACT
Gene identification is an open optimization problem in Bioinformatics. Exponential growth of biological data needs efficient methods for protein translation. Several approaches have been proposed that rely on indicator sequences, statistical and DSP techniques but yet an optimized procedure is required to add an optimal solution. A novel approach for gene identification has been proposed in this paper by employing discrete wavelet transforms for noise reduction in DNA sequences and a novel indicator sequence has been introduced for better signal mapping. Wavelet transforms greatly reduced the background noise and visible peaks of genic regions were found in power spectral estimation. The comparative analysis of proposed and existing approaches showed significant results for novel approach over prevailing solutions for datasets Yersinia pestis (ACCESSION: NC_004088, 4000 bp) and gene F56F11.5 of C elegans (Accession number AF099922) from location 7021. The same significance was observed with four other experiments with real datasets taken from NCBI.
PDF Abstract XML References Citation
Received: September 12, 2010;
Accepted: December 17, 2010;
Published: February 24, 2011
How to cite this article
Muneer Ahmad, Azween Abdullah and Khalid Buragga, 2011. A Novel Optimized Approach for Gene Identification in DNA Sequences. Journal of Applied Sciences, 11: 806-814.
DOI: 10.3923/jas.2011.806.814
URL: https://scialert.net/abstract/?doi=jas.2011.806.814
DOI: 10.3923/jas.2011.806.814
URL: https://scialert.net/abstract/?doi=jas.2011.806.814
INTRODUCTION
Genes on chromosomes are split into two regions i.e., introns and exons (Hamdani and Shukri, 2008 ). Exons are called the coding regions that code for protein. Exon and gene identification is an important task in DNA splicing that lead to better protein translation for consideration to monitor the cell growth, function and type of protein.
Deoxyribonucleic acid (DNA) is a core material in living species responsible for growth and genetic transfer of traits. It is normally found in nuclei of eukaryotic cells (may be found in mitochondrial regions also) contains genes that can be billion of bases long in length. Nucleotide bases are spread over these genes in the form of four important chemical bases, i.e., Adenine (A), Guanine (G), Cytosine (C) and Thymine (T). The bases are always in pairs over DNA ladder aided by a backbone of sugar and phosphate molecules. The sequence and the order in which these bases appear are of fundamental importance for the categorization of variations in acts of species. Diversity in living creatures (mod of behavior) is directly related to degree of differences in organization of bases over chromosomes. The bases may vary from some hundreds to millions and produce the molecules called protein. Protein plays a very fundamental role in growth of different cells and keeping the balance in functional units of body. The replication of DNA for the production of new cells is also an important aspect for revelation of genetic disorders or mutations.
Protein is composed of small scale units called amino acids. There are 20 types of amino acids and the sequence of these units determines the type and function of individual protein molecule.
According to the concepts of Fourier transforms, a signal can be expressed in the form of summation over sine and cosine which only narrates the frequency components of signal (frequency domain analysis) without any depiction of time domain analysis. All frequency components of a digital signal can be obtained but when these components are present and at which time frame (period of time), this information is lacking in Fourier analysis. The restriction is due to inability to cut the signal into pieces and perform the analysis piecewise over the chopped signal. This problem can be stated as Heisenberg uncertainty principal which stated that it is impossible to get the time information of frequency components and also the occurrence of these components in the specified time duration. A more improved solution can be achieved using wavelet transforms.
The gene data is expressed in the form of nucleotides A, T, G, C. indicator sequence methods help us in translation of this data into numeric format that later can be used for spectral analysis of DNA signal. Binary indicator sequence method uses binary values 1 and 0 for the existence or non existence of a specific nucleotide in strand.
In EIIP method, one indicator sequence is proposed as against four binary indicator sequences with numeric values of nucleotides A = 0.1260, T = 0. 1335, G= 0. 0806 and C = 0.1340.
As a replacement of Binary indicator sequence, Complex indicator sequence (Hota and Srivastava, 2008) uses one sequence of values namely X (A) = +1, X (T) = +j, X (G) = -1 and X (C) = -j. The discrete wavelet transform involves the concepts of discritization of continuous transform and discrete coefficients can be calculated using the Eq. 1:
| (1) |
where, a = 2j, b = k2j, j ε N, k ε Z.
The process of performing convolution with scaled wavelet can be repeated so that a set of approximate and detail coefficients can be obtained for each iteration. The discrete transform after normalization can be defined in Eq. 2:
| (2) |
where, k can be termed as a frequency index and alpha as noise index.
Roy et al. (2009) described a generic algorithm for frequency distribution of various spectral values in concern with individual nucleotide bases. Shuo and Yi-sheng (2009) presented an SVM method for prediction accuracy and identification of coding regions.
Chen et al. (2005) proposed a gene prediction system based on Hidden Markov Model (HMM) using Perl and PHP. Guo and Zhu (2008) described a hybrid method comprising Takagi-Sugeno fuzzy model for solution of optimization problem for genic regions identification.
Kakumani et al. (2008) proposed a method by employing statistically optimal null filter for maximization of SNR (signal to noise ratio) and aided with least square optimization criteria. Akhtar et al. (2008a) have shown an optimized solution using Discrete Fourier transforms by monitoring the effect of window lengths for signal processing based coding regions identification. Hota and Srivastava (2008) presented a complex indicator sequence methodology that reduced the computational complexity to 75% than binary indicator sequence method. Akhtar et al. (2008b) have described a DSP method with a comparative analysis of results for proposed and existing solutions. Grandhi and Kumar (2008) have proposed 2-simplex mapping method by assigning the nucleotides to the three corners and one center of a triangle. Mena-Chalco et al. (2008) employed the Modified Gabor-Wavelet Transform for better identification of exons in DNA signal. Gupta et al. (2007) have proposed an approach based on time series analysis. Yin and Yue (2007) have predicted the exonic regions based on period 3 property of exons with implementation of Discrete Fourier transforms and indicator sequence method. Datta and Asif (2005) formulated a Fast Discrete Fourier transform based methodology for genetic regions search in DNA sequence of Eukaryote. Dosay-Akbulut (2006) emphasized the classification of introns in two groups based on RNA secondary structure and self splicing ability in variant species using PCR.
Parent et al. (2004) describe the importance of coordination between transcription and RNA processing that carboxy-terminal domain of RNA polymerase II acts as a common link in both. It highlights two mandatory functions i.e., transcription and later Roxy nucleic acid processing. Coding regions identification helps in smoothing the steps involved in DNA to RNA conversion and drug design.
INDICATOR SEQUENCE
Indicator sequence is used to transform a DNA nucleotide signal (consisting of alphabets A (adenine), G (guanine), Thymine (T) and Cytosine (C)) into some numeric equivalent for revealing the period three component of signal for exonic prediction. The equivalent values of these characters play an important role in discriminating the boundaries between genic and intergenic regions. The indicator sequences proposed in the literature are described below.
Binary indicator sequence: The gene data is expressed in the form of nucleotides A, T, G, C. indicator sequence methods help us in translation of this data into numeric format that later can be used for spectral analysis of DNA signal. Binary indicator sequence method prices 1 and 0 for the existence or non existence of a specific nucleotide in strand.
For example x[n] = [ T T A G G T C C T] translates to [0 0 1 0 0 0 0 0 0] similarly, other binary indicator sequences are formed and then DFT of individual sequences is calculated. Sum of all binary indicator sequences is 1,
uA[n] + uG[n] + uC[n] + uT[n] = 1 |
Let UA[k], UG[k], UC[k] and UT[k] be DFT's of the binary sequences, then:
| (3) |
and Ux may be one of indicator sequences in Eq. 3. After the calculation of DFT:
| (4) |
We need to calculate the absolute value of frequency vector with exponent power 2. This transformation gives us the power spectral density or power spectra of the desired DNA signal described in Eq. 4. The power in the form of magnitude can be plotted against the frequency vector to identify the peaks of exonic regions.
Electron-Ion interaction potential (EIIP) with windowed DFT: In this method, one indicator sequence is proposed as against four binary indicator sequences which computationally reduce the overhead by 75%:
YEIIP = WAXA + WTXT + WCXC + WGXG |
where, numerical values are:
A = 0.1260 |
And the transform becomes:
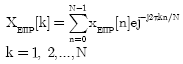 | (5) |
where, k is bound in sample space, 0 ![]() k
k ![]() N
N
Complex indicator sequence with windowed DFT: As a replacement of binary indicator sequences, complex indicator sequence uses one sequence of values (Hota and Srivastava, 2008) namely:
| X (A) = +1 |
And the corresponding transform becomes Eq. 6:
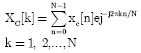 | (6) |
where, value of k remains between the sample space bounds.
The method of Complex Indicator Sequence reduces the computational overhead by 75% and provides more accurate prediction of genic regions.
DIGITAL FILTER METHODS
Finite impulse response filter (FIR): The filters that carry a finite response to impulse signals are called FIR filters. The FIR filter of length k can be described as:
| (7) |
where, Y is the transformed data and x is the input data. The filter takes a summation over input vector multiplied by a constant factor. The output vector has the same length as input vector. K is called the order of this filter:
where, A (z) is a transfer function for this filter. It is obtained by dividing the output vector values by the input vector. We can also term this as:
Which shows a polynomial equation in z-transform and defines the same FIR filter? These filters are widely used because of their stability.
Infinite impulse response filter (IIR): This filter carries an infinite response to signal:
| (8) |
where, y represents a vector of length n that contains the transformed values for IIR filter. The filter used two kinds of coefficients, feed forward and feed backward represented by ak and bk:
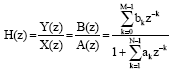 | (9) |
where, H is the transform function over z-transform when output vector is divided by input vector. The main difference between the two filters is stability, band width and order of filter. IIR filter with its extension is widely used in DSP techniques for DNA signal analysis.
DISCRETE WAVELET TRANSFORMS
Discrete Wavelet transforms provide the best time scale localization of DNA signal. We have used DWT for denoising our signals.
A Wavelet transform can be presented as:
| (10) |
where, Ψ(t) is mother wavelet and b is shift parameter, the Discrete coefficients after choosing values of a (initial) =2 and b (initial) = 1 can be written as:
| (11) |
PROPOSED APPROACH
Our hybrid approach contains the following components:
| • | Employing indicator sequence |
| • | Noise reduction |
| • | Segmentation in frames |
| • | PSD estimation |
| • | Discrimination measure estimation |
| • | Nucleotide range estimation |
Mapping: For mapping the nucleotides in gene sequence to a DNA signal, we have introduced a novel indicator sequence (called UTP, University Technology PETRONAS indicator sequence) after a keen analysis of nucleotides in codon clusters of coding regions in DNA signals of huge datasets. The numeric equivalents of nucleotides for this indicator sequence are stated as Adenine (A) = X (A) = 0.260, Thymine (T) = X (T) = 0.375, Guanine (G) = X (G) = 0.125 and Cytosine (C) = X (C) = 0.370.
Noise reduction: We have used Daubechies Discrete Wavelet transforms for denoising our DNA signal of Yersinia by setting the appropriate frequency component thresholds in analysis of approximate and detail coefficients. These coefficients corresponded to the low and high scale frequency components of signal.
Figure 1 describes the down-sampling and up-sampling of DNA signal of Yersinia by Daubechies transforms of order three. The signal is passed through filters of low scale (high frequency) and high scale (low frequency) for generation of vectors containing approximate and detail frequency components information of signal. These vectors contain the half of the signal information each. First level coefficients of high pass are buffered while low pass coefficients are again down-sampled by a factor of 2. Second level coefficients of high pass are again buffered and low pass components are down-sampled again for third level coefficient generation. The third level frequency components of high pass signal are buffered and signal is decomposed. The same process is applied with the help of low and high pass filters in up-sampling. The denoising of DNA signal for Yersinia helps in appropriate estimation of discrimination factor for exons and suppression of 1/f noise.
Segmentation in frames: We have found Kaiser Window of length 351 bp:
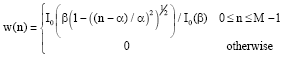 |
with β = 3.5 (minimizes the leakage factor and enhances the main lobe width) after careful and thorough analysis of variant functions combinations with different parametric values.
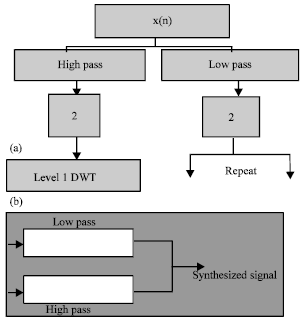 | |
| Fig. 1: | Daubechies wavelet transforms (a) down-sampling (b) up-sampling |
Magnitude and power measures of frames: Magnitude and power of each frame is calculated and frequencies are normalized for better PSD estimation:
|Frame| = Ax(f) = | Xl(f)| (Magnitude of frame) |
Also called absolute value
Power of Frame = Absolute value of frame raised to the power of factor 2 = |Frame|2 = Px (f) = |Xl(f)|2
The frequency components are then normalized by:
where, fs is the sampling frequency and L is the length of original signal.
It is worth mentioning that the any increment in normalization factor beyond fs*L creates a need for rescaling the frequency vector rather than any further improvement in spectral analysis.
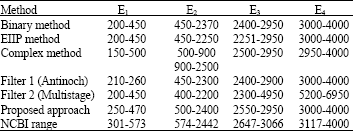
Discrimination measure estimation: Discrimination measure is a ratio of lowest exonic peak height (in a set of exons) to the heights peak value of intron (in a set of introns) in estimation of power spectral density of frames. The calculated discrimination measure for proposed and existing approaches is shown in Table 1 under results and discussions.
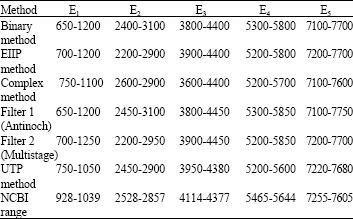
Nucleotide range estimation: The genic regions bounds are estimated from power spectral density estimation plots. The results for exonic boundaries for specie Yersinia with 4000 bp have been summarized in Table 2.
RESULTS AND DISCUSSION
We have used dataset Yersinia pestis (ACCESSION: NC_004088, 4000 bp, that contains four genes and exons from location 5000 to 8999 bp) for comparative analysis of coding regions identification. Significant improvement in prediction was obtained in calculation of discrimination measure for PSD estimation in proposed and all existing approaches. The prevailing methods include Binary indicator sequence method (Anastassiou, 2001) EIIP method (Achuthsankar and Sivarama, 2006), Complex indicator sequence method (Hota and Srivastava, 2008) Digital filter methods (Vaidyanathan and Yoon, 2002; 2004) .
Figure 2a narrates the PSD for Binary indicator sequence method (Anastassiou, 2001). There is a considerable difference of bounds 100 bp almost for nucleotide ranges than NCBI. The third exon carries promising difference of 200 bp for the first initiation with a slight difference in terminating region. The EIIP method (Achuthsankar and Sivarama, 2006) in Fig. 2b shows the same behavior for the first and second gene but there is a variation in nucleotide range for third exon. Third exon carries a major gap of almost 400 bp which is obviously another revealing flip for this method.
Figure 2c describes Complex indicator sequence method (Hota and Srivastava, 2008). The first gene carries the major gap in nucleotide ranges than NCBI standard range (almost 150 bp in initiation). There is a breakup of range for the second exon between 500-900 and then 900 to 2500 bp. 3rd gene is more close to the standard range than Binary and EIIP methods. The proposed approach in Fig. 2d describes the more promising close range of nucleotide to the standard range. We can see a clear difference of closeness of bounds compared with the prevailing methods.
Table 1 describes the exonic boundaries calculated against different approaches. Complex method contains a disconnection in second exon and exon peaks are far from the standard range. The proposed approach bestows the closer range comparable with NCBI range.
Table 2 presents the comparative analysis of proposed and existing approaches for discrimination measure. We can see a larger value for this factor for proposed approach. There is a gain of 100% than Filter 2 (Vaidyanathan and Yoon, 2002) 114% than Filter 1 (Vaidyanathan and Yoon, 2004) 20% than Complex indicator sequence method, 159% than EIIP method an 138% than Binary method. This significant improvement in results depicts the outperformance of proposed approach.
| Table 1: | Exon Boundaries in different approaches |
 | |
| Table 2: | Comparative analysis of various methods |
 | |
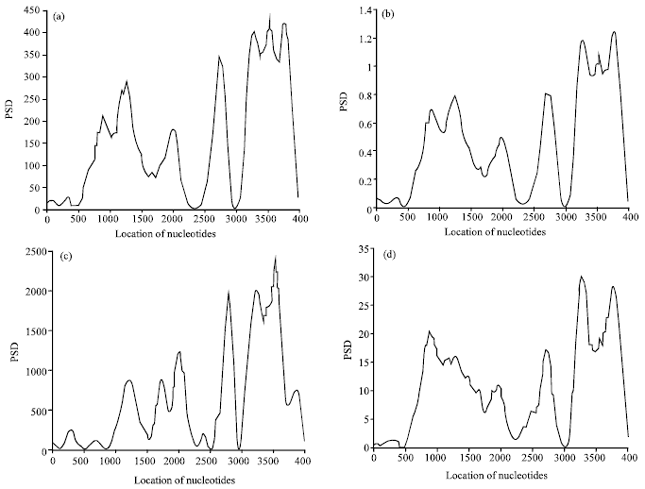 | |
| Fig. 2: | Power spectral density estimation methods (a) Binary; (b) EIIP; (c) Complex and (d) Proposed |
 | |
| Fig. 3: | PSD of complex versus proposed method |
Figure 3 shows the PSD of Complex method against the proposed approach. X and Y axis represent the nucleotide locations and power spectral density estimation respectively. We can monitor larger exon peaks of proposed approach against the promising Complex indicator method (Hota and Srivastava, 2008) with discrimination measure of 2.5. The discontinuity seen in Complex method for second exon was removed in proposed method. The third exon contains a larger peak in Complex indicator method while peaks for the fourth exons are almost similar. First exon carries high peak against a comparatively high peak of intron in Complex indicator method.
Power spectral analysis over S.cerevisiae dataset: The power spectral analysis for gene F56F11.5 of C elegans (Accession number AF099922) from location 7021 is depicted in Fig. 4.
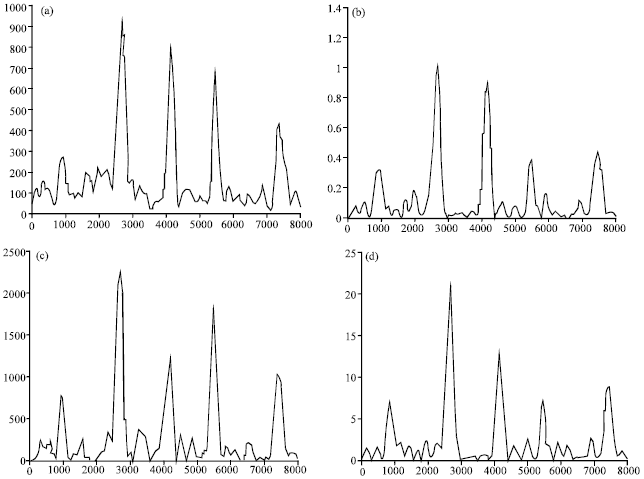 | |
| Fig. 4: | Power spectral density estimation methods (a) Binary; (b) EIIP; (c) Complex and (d) Proposed |
All sections of Fig. 4 depict various methods employed for spectral analysis of gene S. cerevisiae chromosome III (AF099922). X-axis and Y-axis represent the nucleotides and PSD respectively. We have calculated the Discrimination factor D for all methods. The Discrimination factor is the ratio of lowest peak in set of exonic peaks to the highest peak in set of intronic peaks. Greater the value of D, greater is the prediction accuracy and clear differentiation can be made between introns and exons. Numeric value of D is another picture of minimization of 1/f noise and maximization of genic peak values.
Table 3 describes the comparative analysis of various methods for power spectral analysis performed over S.cerevisiae chromosome III. We can see that Complex indicator sequence method (Hota and Srivastava, 2008) generates D as 2.06 which was the highest discriminant value over all existing techniques. The UTP indicator sequence with wavelet transforms generates D as 2.8 which provide 36% more prediction accuracy.
We obtained a gain of 130% prediction accuracy than Filter 2, 166% than Filter 1, 65% than EIIP indicator sequence method and 133% than Binary indicator sequence method (Anastassiou, 2001).
We calculated the nucleotide range for exons and summarized as follows:
Table 4 summarizes the nucleotide range of five exons. We can monitor clear differences as a comparative analysis of various approaches. Binary and EIIP methods glimpse more or less wide range difference than standard NCBI results. Complex method results are better than the first two approaches. Filter 1 and 2 behave accordingly while there is significant improvement in prediction of exons range with proposed approach.
Results and discussion section reveals that period-3 property is more significant in proposed approach. We have measured the discrimination measure in power spectral estimation using six real datasets (results for two datasets have been included here) from NCBI. Comparing the results mentioned in all tables, we found high sharp genic peaks and reduced background 1/f noise in proposed approach. These results have been explained using appropriate figures for PSD's and tables for discrimination measure estimation in both respects (maximization of peaks and minimization of DNA sequence noise).
| Table 3: | Numerical evaluation of discrimination measure |
 | |
| Table 4: | Nucleotide range for exons |
 | |
The calculations for bounds and peaks have been discussed in favor and contradiction of proposed technique against existing solutions. The trade off between DNA sequence noise and peak heights has been minutely described in the form of discontinuity in graphs and nucleotide range estimation.
CONCLUSION
A novel method for gene identification is proposed in this paper. The method reduces the background noise in DNA signal by employing the discrete wavelet transforms along with mapping nucleotide with a new indicator sequence. The power spectral analysis and discrimination measures calculated over Yersinia pestis (ACCESSION: NC_004088, 4000 bp) and gene F56F11.5 of C elegans (Accession number AF099922) from location 7021 showed significant improvement in coding regions identification compared with existing techniques. The computational overhead is also reduced to 75% than traditional Binary indicator method. The significant improvement in prediction may help in understanding cell growth, function and protein transcription and drug design. The same significance was observed with four other real datasets.
ACKNOWLEDGMENT
We are obliged for the kind assistance and support from Departments of Computer Sciences at King Faisal University Saudi Arabia and University Technology Petronas Malaysia.
REFERENCES
- Roy, M., S. Biswas and S. Barman, 2009. Identification and analysis of coding and noncoding regions of a DNA sequence by positional frequency distribution of nucleotides (PFDN) algorithm. Proceedings of the 4th International Conference on Computers and Devices for Communication, December 14-16, 2009, Kolkata, India, pp: 1-4.
Direct Link - Guo, S. and Y.S. Zhu, 2009. Prediction of protein coding regions by support vector machine. Proceedings of the International Symposium on Intelligent Ubiquitous Computing and Education, May 15-16, 2009, Chengdu, China, pp: 185-188.
CrossRefDirect Link - Hamdani, H.Y. and S.R.M. Shukri, 2008. Gene prediction system. Proceedings of the International Symposium on Information Technology, Volume 2, August 26-28, 2008, Kuala Lumpur, Malaysia, pp: 1-7.
CrossRefDirect Link - Guo, S. and Y.S. Zhu, 2008. An integrative algorithm for predicting protein coding regions. Proceedings of the IEEE Asia Pacific Conference on Circuits and Systems, November 30-December 3, 2008, Macao, pp: 438-441.
CrossRefDirect Link - Kakumani, R., V. Devabhaktuni and M.O. Ahmad, 2008. Prediction of protein-coding regions in DNA sequences using a model-based approach. Proceedings of the IEEE International Symposium on Circuits and Systems, May 18-21, 2008, Seattle, WA., USA., pp: 1918-1921.
CrossRef - Akhtar, M., E. Ambikairajah and J. Epps, 2008. Optimizing period-3 methods for eukaryotic gene prediction. Proceedings of the IEEE International Conference on Acoustics, Speech and Signal Processing, March 31-April 4, 2008, Las Vagas, NV., USA., pp: 621-624.
CrossRef - Hota, M.K. and V.K. Srivastava, 2008. DSP technique for gene and exon prediction taking complex indicator sequence. Proceedings of the IEEE Region 10 Conference, November 19-21, 2008, Hyderabad, India, pp: 1-6.
CrossRefDirect Link - Akhtar, M., J. Epps and E. Ambikairajah, 2008. Processing in sequence analysis: Advances in eukaryotic gene prediction. IEEE J. Signal Selected Topics Signal Proc., 2: 310-321.
Direct Link - Grandhi, D.G. and C.V. Kumar, 2008. 2-Simplex mapping for identifying the protein coding regions in DNA. Proceedings of the IEEE Region Conference on TENCON, October 30-November 2, 2008, Tiapei, pp: 1-3.
CrossRef - Mena-Chalco, J.P., H. Carrer, Y. Zana and R.M. Cesar, 2008. Identification of protein coding regions using the modified gabor-wavelet transform. IEEE/ACM Trans. Comput. Biol. Bioinform., 5: 198-207.
CrossRef - Gupta, R., A. Mittal, K. Singh, P. Bajpai and S. Prakash, 2007. A time series approach for identification of exons and introns. Proceedings of the 10th International Conference on Information Technology, Dec. 17-20, India, pp: 91-93.
Direct Link - Datta, S. and A. Asif, 2005. A fast DFT based gene prediction algorithm for identification of protein coding regions. Proceedings of the IEEE International Conference on Acoustics, Speech and Signal Processing, March 18-23, USA., pp: 653-656.
Direct Link - Chen, H., F. Gu and F. Liu, 2005. Predicting protein secondary structure using continuous wavelet transform and Chou-Fasman method. Conf. Proc. IEEE Eng. Med. Biol. Soc., 3: 2603-2606.
PubMed - Dosay-Akbulut, M., 2006. Group I introns and splicing mechanism and their present possibilities in elasmobranches. J. Biol. Sci., 6: 921-925.
CrossRefDirect Link - Parent, A., I. Benzaghou, I. Bougie and M. Bisaillon, 2004. Transcription and mRNA processing events: The importance of coordination. J. Biol. Sci., 4: 624-627.
CrossRefDirect Link - Vaidyanathan, P.P. and B.J. Yoon, 2004. The role of signal processing concepts in genomics and proteomics. J. Franklin Instit., 341: 111-135.
CrossRef - Achuthsankar, S.N. and P.S. Sivarama, 2006. A coding measure scheme employing Electron-Ion Interaction Pseudopotential (EIIP). Bioinformation, 1: 197-202.
Direct Link - Vaidyanathan, P.P. and B.J. Yoon, 2002. Gene and exon prediction using allpass-based filters. Proceedings of the Workshop on Genomic Signal Processing and Statistics, October 2002, Raleigh, NC, USA., pp: 1-4.
Direct Link