
Safeia Hamasha
Department of Physics, Faculty of Science, Hashemite University, P.O. Box (150459), Zarqa 13115, Jordan
Mahmoud Abu Allaban
Department of Physics, Faculty of Science, Hashemite University, P.O. Box (150459), Zarqa 13115, Jordan
Amani Tahat
Faculty of Natural Resources, Hashemite University, P.O. Box 150459, Zarqa 13115, Jordan
Journal of Applied Sciences
Year: 2011 | Volume: 11 | Issue: 15 | Page No.: 2686-2699







ABSTRACT
In this study, a new atomic physics program (HTAC) is introduced and tested. It is a utility program designed to automate the computation of various atomic structure and spectral data. It is the first comprehensive code that enables performing atomic calculations based on three advanced theories: the fully relativistic configuration interactions approach, the multi-reference many body perturbation theory and the R-Matrix method. It has been designed to generate tabulated atomic data files that can be interfaced with other programs in a simple and straightforward manner. It works for all known elements in the periodic table and their ions. Generated atomic structure data includes energy levels, oscillator strength, radiative transition rates, mixing coefficients for any rank of multipole transitions and other quantities. It provides a user friendly interface that works under windows in the dialog mode. Its Graphical User Interface (GUI) clarifies analysis steps and handles file manipulations required by the used model. In addition, the GUI provides viewing and plotting capabilities of the generated atomic data. Furthermore it will be distributed freely.
PDF Abstract XML References Citation
Received: February 16, 2011;
Accepted: May 21, 2011;
Published: July 02, 2011
How to cite this article
Safeia Hamasha, Mahmoud Abu Allaban and Amani Tahat, 2011. Developing a New Atomic Physics Computer Program (HTAC) to Perform Atomic Structure and Transition Rate Calculations in Three Advanced Methods. Journal of Applied Sciences, 11: 2686-2699.
DOI: 10.3923/jas.2011.2686.2699
URL: https://scialert.net/abstract/?doi=jas.2011.2686.2699
DOI: 10.3923/jas.2011.2686.2699
URL: https://scialert.net/abstract/?doi=jas.2011.2686.2699
INTRODUCTION
Theoretical atomic data is important for many physics research fields including plasma modeling under various physical conditions, X-ray spectroscopy and astrophysics research. In order to respond to the need of atomic data, scientists have developed several computer programs that can perform atomic calculations. Some atomic codes such as CIV3 (Hibbert, 1975; Gupta and Msezane, 2009), super structure (Eissner et al., 1974; Del Zanna et al., 2005) and COWAN code (Cowan, 1981) are based on the non-relativistic approximations whereas the RMBPT code is based on a relativistic many body perturbation theory that includes the first and the second order relativistic energy corrections (Safronova et al., 2006; Safronova et al., 1996), HULLAC (Busquet et al., 2006), SZ (Zhanget al., 1989) and ATOM (Amusia and Chernysheva, 1997) are fully relativistic atomic codes developed based on solving Dirac equations. The Flexible Atomic Code (FAC) is a fully relativistic code that combines the strengths of the available codes with modifications to numerical methods (Gu, 2008). FAC is available for the public at no cost and can be downloaded from the web (http://sprg.ssl.berkeley.edu/~mfgu/fac/fac.tar.gz). FAC itself is a powerful computation code but it is written in Python programming language which is not famous in the atomic physics community. Therefore, utilizing FAC to generate atomic data has been limited to its author in addition to few researchers who are familiar with python language.
Available atomic physics codes are often complicated, difficult to use and most of them works under UNIX or other operation systems that work through command typing.
This research is a step forward to simplify atomic calculations by introducing Hamasha-Tahat Atomic Code (HTAC). HTAC is a comprehensive code that enables performing atomic calculations based on three advanced theories: the fully relativistic configuration interactions approach, the multi-reference many body perturbation theory and the R-Matrix method. It has a user friendly interface enables researchers to use it without going through the complications of atomic theories or the programming languages. HTAC will be available for public at no charge upon their request.
In developing HTAC, several functions and subroutines that perform atomic calculations through the fully relativistic Configuration Interaction Method (CIM) from FAC were used though its binary files. In addition, the multi-reference many body perturbation theory (MR-MBPT) outlined by Vilkas et al. (1998) was also used and The R-Matrix method is impeded in HTAC too. Several Free software including gcc, gfortran (c/c++/ff77 compiler,tk/tcl interpreter were obtained from www.cygwin.com.
This study presents the atomic structure program part which constitutes the first component in HTAC while other components are being developed in order to come up with a complete package of advanced atomic programs that performs atomic calculations based on advanced atomic theories.
MATERIALS AND METHODS
Fully relativistic configuration interaction approach (RCIM): This method follows the flexible atomic code methodology. In this section the theory of the fully Relativistic Configuration Interaction Approach (RCIM) is briefly described. The bound states of the system are calculated in the configuration mixing approximations with convenient specification of the mixing scheme. The radial orbitals for the construction of the basis states are derived from the modified self-consistent Dirac-Fock-Slater iteration on a fictitious mean configuration with fractional occupation numbers representing the average electron cloud of the configurations included in the calculation.
In atomic units, the relativistic Hamiltonian (H) of an ion with N electrons is given by:
| (1) |
where, HD (I) is the single electron Dirac Hamiltonian of a potential attributed to the nucleus charge which should be diagonolized in order to obtain energy levels.
The approximate atomic state functions are given by:
where, bμ are the mixing coefficients obtained from the diagonalization of the total Hamiltonian and φμ are the bases states which are anti-symmetric sums of the products of the N Dirac spinors (φnkm) that are given by:
where, Pnk and Qnk are the large and small components, respectively; χkm is the spin angular function, n is the principle quantum number, k is the relativistic angular momentum which is equal to (l-j) (2j+1), m is the magnetic quantum number l is the orbital angular momentum and j is the total angular momentum.
Pnk and Qnk satisfy the coupled Dirac equation for central field V (r):
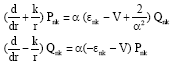 | (2) |
where, α is the fine structure constant and εnk is the energy eigenvalues of the radial orbitals. V (r) is the local central potential which is the sum of the nuclear charge contribution potential (VN (r)) and the electron-electron interaction potential (Vee (r)).
VN (r) is given by:
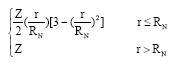 | (3) |
where, RN is the statistical model radius of the nucleus calculated from the formula RN = 2.02677x10-5 A1/3 where A is the atomic weight (Chernysheva and Yakontov, 1999). The electron-electron interaction approximation is given by the following equation which is corrected at the asymptotic behavior (large r) by excluding the self- interaction term:
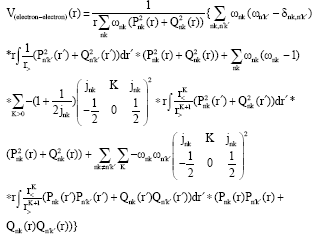 | (4) |
where:
is the Wigner 3-j symbol and r< and r> are the less or grater of r and r�. The electron-electron interaction contribution to the average energy Ee-e is given by:
| (5) |
The factor ½ is introduced in order to avoid double counting of electron pairs in the summation.
Equation 2 is solved by constructing a self-consistent iteration, in which the orbital wave function from the previous step is used to derive the potential. It is converted to a Schrödinger-like equation by eliminating the small component and performing the following transformation (Chernysheva and Yakontov, 1999):
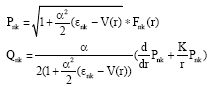 | (6) |
Equation 3 is rewritten as:
| (7) |
where, U (r) is an effective potential defined as:
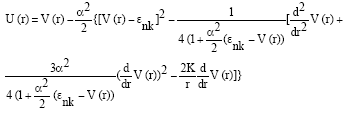 | (8) |
Another transformation may be performed:
| (9) |
| (10) |
Therefore, Eq. 7 becomes a Schrödinger-like form given by:
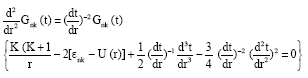 | (11) |
The used form of t (r) is:
| (12) |
This form covers larger radial distance than a linear form for a given number of grid points does. The standard Numerov method is used to solve the Schrödinger like equation after the performed transformations (Eq. 11). The minimum and maximum distances on the radial grid are chosen to be within the nuclear charge distribution and the excited states to be below shell number 20 and the bound energies are less than the Coulomb potential at rmax such that: rmin = 10-6/Zeff and rmax = 500/Zeff, Zeff is the effective charge of the ion that the electron experiences at large r.
The radiative transition rates are calculated in a single multipole approximation where the multipole operator is: ![]() , the initial state is:
, the initial state is:
and the final state is:
The second quantization method is used to solve the Hamiltonian matrix elements by recoupling the creation and annihilation operators with the help of Racah algebra. By introducing the Wigner-Ekraft theorem, the tensorial multipole operator Eq. 15 can be written as:
| (13) |
where,![]() denotes the reduced matrix element and α, β denotes only the states with quantum numbers nk because the summation over m is contained in:
denotes the reduced matrix element and α, β denotes only the states with quantum numbers nk because the summation over m is contained in:
The matrix elements are obtained following Gaigalas et al. (1997). The line strength of the transition is:
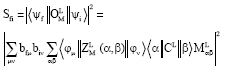 | (14) |
where, ![]() is the radial part of the single electron multipole operator.
is the radial part of the single electron multipole operator.
The weighted oscillator strength is given by:
| (15) |
The weighted transition rates are given by:
| (16) |
where, ω = Ef-Ei is the transition energy.
The multi- reference many body perturbation theory approach: The numerical procedure of this method follows Vilkas et al. (1998, 2006) and Ishikawa et al. (2010). Starting with Rayleiigh- Schrödinger perturbation theory for a multi-configurational model space.
| (17) |
where, HDCB is Dirac-Coulomb- Breit (DCB) Hamiltonian for an N-electron ionic system given by:
| (18) |
where, hd (i) is the Dirac Hamiltonian for one free electron. ri is the radial coordinate of the electron i. rij is the distance between the electrons i, j and Bij is the frequency-independent Breit interaction:
| (19) |
| (20) |
where, σi is the Pauli spin matrices.
The DCB Hamiltonian is covariant to first order. It improves the accuracy of calculating the fine structure splitting and the inner shell binding energies.
We may write: HDCB = H0+V where:
| (21) |
| (22) |
The screening effect of all electrons is included in the potential U (r) which is chosen with careful attention in order to make the perturbation potential (V) as small as possible. U (r) is approximated by a local central potential derived from a Dirac-Fock-Slater self-consistent field calculation by minimizing the weighted mean energy of the configuration within a given group. Determining U (r) allows the determination of the eigenvectors and eigenvalues of H0 by forming a Slater determinants from single electron wave functions (Φk) of the Configuration State Functions (CSF) such that
The atomic state function Ψk that belongs to the model space M is defined as a linear combination of the basis function Φk.
| (23) |
where, cki are the mixing coefficients determined from the eigenvalues by diagonalizing the Configuration Interaction (CI) matrix.
The multi-reference many body perturbation theory (MR-MBPT) is used to determine energy levels. The initial step for MR-MBPT is to determine a set of Dirac spinors via the configuration interaction method (multi-configuration-Dirac-Fock-Breit self-consistent field calculations) for a many electron system. The eigenfunctions (![]() ) are obtained by diagonalizing DCB Hamiltonian which is partitioned into two subspaces; M and N. Subspace M consists of configurations included in the CI expansion whose correlations are treated to all orders. Subspace N contains the residual dynamic correlations that are subjected to the second order many body perturbation theory.
) are obtained by diagonalizing DCB Hamiltonian which is partitioned into two subspaces; M and N. Subspace M consists of configurations included in the CI expansion whose correlations are treated to all orders. Subspace N contains the residual dynamic correlations that are subjected to the second order many body perturbation theory.
The energy levels are given by:
| (24) |
where, ![]() is the CI eigenvalues and
is the CI eigenvalues and ![]() is the second order correction due correlations in the subspace N.
is the second order correction due correlations in the subspace N. ![]() is given by:
is given by:
| (25) |
where, ![]() and
and ![]() are the zeroth order energies of the configuration state functions.
are the zeroth order energies of the configuration state functions.
The large and small components of Dirac spinors should be chosen to satisfy the boundary conditions of the finite nucleolus and the boundary radius (Q (rb)/P(rb) = b). where, rb is the boundary radius which is chosen to be large enough to contain all amplitudes of the wave function. The arbitrary constant b is chosen to be κ/2rbc where κ is the relativistic angular momentum quantum number and c is light speed in space.
The first order MR- MBPT wave functions are employed to calculate transition rates which is expressed in terms of CSFs:
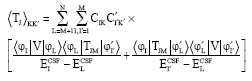 | (26) |
where, L is over intermediate states.
R-matrix theory: R-matrix method is a calculational tool based on scattering theory (Ekuma, 2007) by deriving scattering properties from the Schrödinger equation in a large variety of physical problems. Its principle relies on a division of the configuration space into two regions: the internal and external regions. The boundary between these regions is defined by a parameter known as the channel radius. The details may be found in many sources as in Descouvemont and Baye (2010).
Running atomic structure program: Figure 1 shows HTAC structure with the extensions of their output files along with the three calculation methods. “*. LEV” is the extension for the energy levels tables.“*.TR ” is the extension of transition rates tables. “*.LEV.MC” for the mixing coefficients of the energy levels table and “*.WV” for the wave functions details table. The method of calculation for MBPT and R-matrix could be denoted from the output file extension. S and D in the many body perturbation method (MBPT) denotes single and double excitation, respectively. But the default method is RCIM has the formal extensions names of the output tables.
The program works in a dialog mode. Once HTAC is installed in the personal computer, it can be run easily through Windows by clicking its icon which might be placed at the desktop. Atomic calculations are initiated by pointing the "Atomic Calculations" button in HTAC standard tool bar which opens the atomic structure main panel (Fig. 2). The atomic structure program allows defining the input data needed for calculating atomic structure and transition rates in simple ways. Input file includes the element to be studied, its ground electronic configuration and the electronic configurations of the initial and final states of the transition. User has three ways to specify input data files:
| • | Default data is provided through a built-in input file named “structure.in”. It contains data required to obtain energy levels and radiative transition rates for Ne-like iron ion which has the ground state electronic configuration 1s2 2s2 2p6. The transition occurs between groups (or complexes) n = 2 and n = 3 and leads to the final configuration which is described by: 1s2 2s2 2p5 3* 1. 1s and 2s are closed shells, therefore a single multipole transition occurs between 2p sub-shell to any n=3 sub shell according to the multipole transition selection rules. The input file contains the element to be studied, number of electrons, closed shells and information about initial electronic configuration (before transition) and final electronic configuration (after transition). The user is encouraged to refer to the tutoring example which can be accessed through the "help" button in HTAC main panel of atomic structure program (Fig. 3) |
| • | A comprehensive built-in database for 100 elements and their characteristics including atomic number, closed shell, initial configuration, final configuration and other important information. HTAC user may import input files for any element through the "Open" button in the file menu |
| • | User may build up his/her own input file by selecting "clear" button which erase all fields in the main panel. After that the user fills blank spaces and save the constructed input file through the "save as" button. The user may restore the default values at any stage |
HTAC atomic structure program provides three options to the structure function based on three theoretical methods; the fully relativistic configuration interaction (RCIM) which is the default method, the Many Body Perturbation Theory (MBPT) and R-matrix. User can switch to MBPT method or the R-Matrix method by the tab option. Both methods have different panels (Fig. 4). Output options of MBPT includes; a single excitation transition rates output, a double excitation transition rates output and a combined (single and double) rates output. Another output MBPT option is to request produced data in a length form (Babushkin gauge) or a velocity form (Coulomb gauge) (Ishikawa et al., 2010). All methods are used to calculate transition rates for all optically allowed and forbidden transitions. User can choose the rank (k) of any electric multipole transition (Ek) or the rank of any magnetic multipole transitions (Mk).
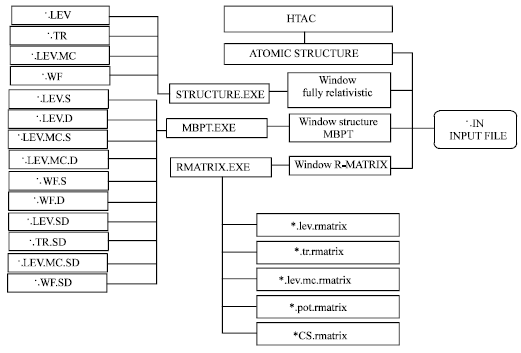 | |
| Fig. 1: | The structure of HTAC atomic structure program with the three calculation methods and the extensions of their output files |
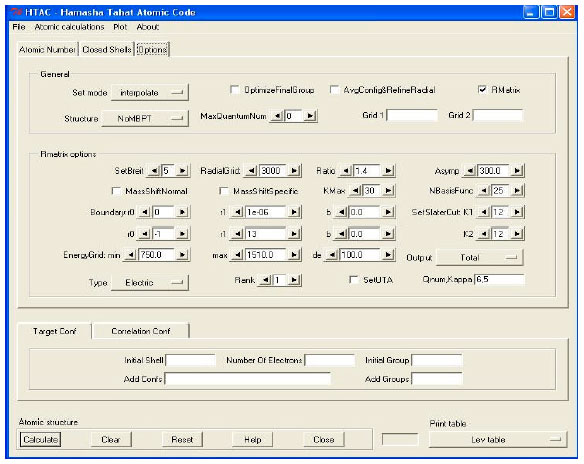 | |
| Fig. 2: | The Main panel of HTAC atomic structure program |
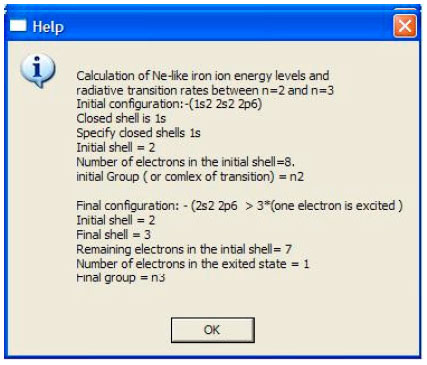 | |
| Fig. 3: | The help window demonstrates an example for Ne-like iron ion input-file |
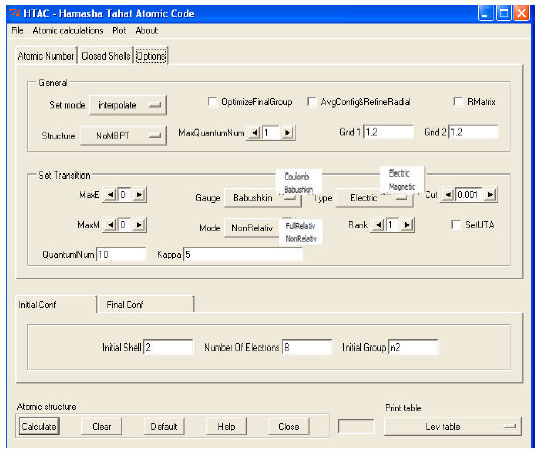 | |
| Fig. 4: | The MBPT panel with its options |
There are four output files for CIM, MBPT and R-Matrix methods:
| • | The energy level table: It contains the atomic number, the ground state energy in eV, the energy of excited levels with respect to the ground state energy in eV, states parity, 2j and the electronic configuration of each level |
| • | The transition rates tables: It contains the atomic number, 2j, deltaE, the oscillator strength, the transition rate and the electric and magnetic multipoles reduced matrix elements |
| • | The mixing coefficients table: It contains the basis table (p, j, state configuration, level names) and the mixing coefficients level symmetry |
| • | The wavefunctions table: It contains the quantum number, Kabba, the energy and details of the wave function (Fig. 5). Used units are sec-1 for transition rates and eV for energies |
R_MATRIX parameters: The R-matrix parameters appear in Fig. 1 includes (note: the parameters are written as they appear in the program):
| • | SetBreit counter; it sets maximum principle quantum number of the states, for which the Breit interaction should be included in the Hamiltonian. It accepts any integer between zero and ten. If SetBreit counter is assigned a negative value, its interaction will include all bound and continuum states |
| • | NBasisfunc counter; it represents allowed number of the basis functions per state. It takes values between zero and hundred |
| • | KMAX counter; it represents the maximum orbital angular momentum (k) of the continuum electron. It takes values between zero and hundred |
| • | Boundary counters (r0, r1, b); r0 accepts values between -100 and 100, r1 from zero to 100 and b takes values between 0 and 1000. If r0 is assigned any value other than zero, the code sets the boundary conditions in the propagation zone |
| • | SetSlaterCut counter (K1, K2); it allows determination of the values of the orbital angular momenta k1 and k2 which determine the calculation modes of Slater integrals. If the orbital has an angular momentum greater than the assigned k1 value, the exchange integrals will not be calculated. If the angular momentum is greater than the assigned value of counter k2, the direct integrals will be evaluated along with the reduced multipole matrix elements |
| • | RadialGrid counter; it allows setting the radial grid properties |
| • | Ratio counter; it allows specifying the ratio of successive radial points near the origin which is approximately logarithmic |
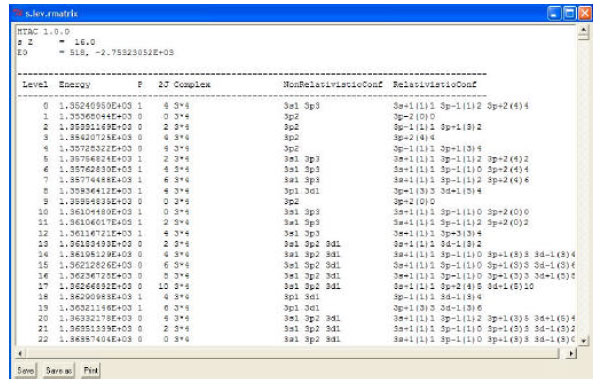 | |
| Fig. 5: | The energy levels output file for Sulfur (S. Z = 16) saved under file name "s.lev.rmatrix" |
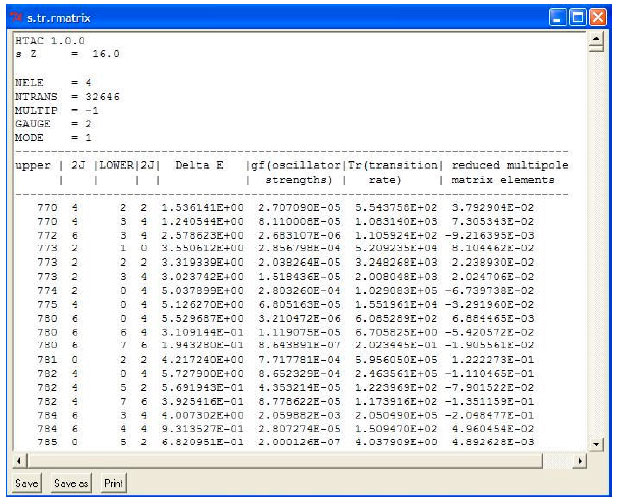 | |
| Fig. 6: | Electric dipole (E1) radiative transition table produced by using R-MATRIX method for Sulfur saved under file name "s.tr.rmatrix" |
| • | Energy Grid and related counters; it allows specifying the energy grid |
| • | Asymp counter; it allows setting the number of terms in the Gailitis asymptotic expansion |
| • | MassShiftNormal and massshiftpecifi check boxes; they allow setting flags for normal mass shift and specific mass shift contributions to the Hamiltonian. They are enabled/ disenabled by pointing |
| • | Type menu; it allows setting the type of electric- or magnetic- multipole transitions |
| • | Rank counter; it allows setting the multipole transition rank |
| • | Default values for above functions appear once the R-matrix check box is clicked. Examples of output files of energy levels and transition rates produced via the R-matrix option in HTAC are presented in Fig. 5 and 6 |
Warning messages: In order to minimize personal errors, HTAC warns the user of any committed mistake by displaying a warning massage window contains detailed information that leads the user to the error. This option is particularly important to save time and to ensure high quality of the produced atomic data. Figure 7 shows two examples of warning message as displayed by HTAC.
Application: Atomic structure, electric multipole transitions (E1, E2, E3) and magnetic multipole transitions (M1, M2, M3) atomic data of Ne-like Fe ion: Here, a real example of generation atomic structure and transition rates of allowed and forbidden transitions data is presented. The chosen ion has a great interest in astrophysics which is Ne- like iron ion (Fe16+). Please note that the output files for the energy levels and the transition rates generated by all methods would have similar table format.
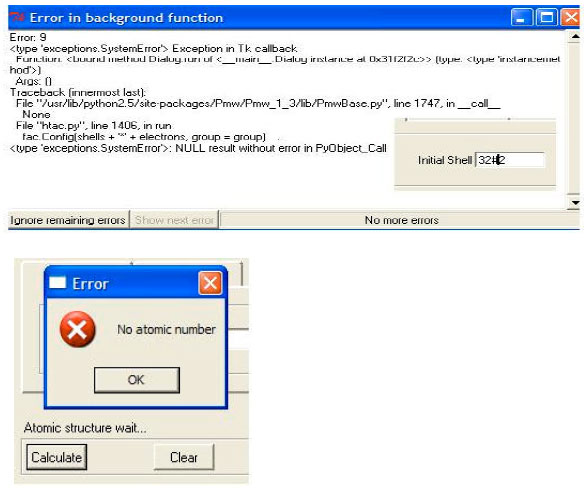 | |
| Fig. 7: | Warning messages pup up when a mistake is committed |
RESULTS
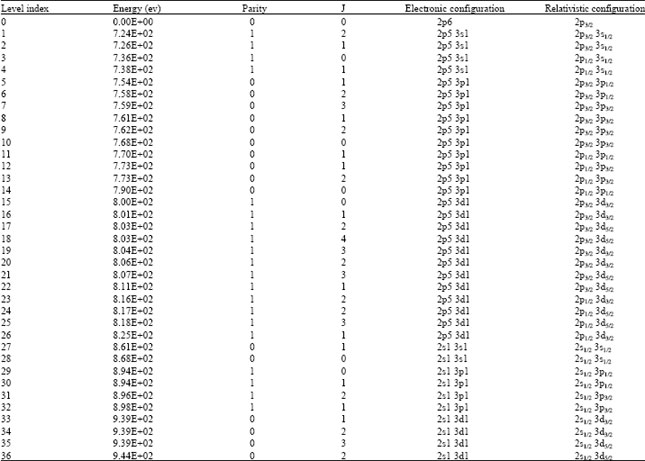
Fully relativistic configuration interaction method: The ground state electronic configuration for Ne- like Fe (Fe16+) is 1s2 2s2 2p6. Calculated data for atomic structure of Ne-like Fe is shown in Table 1. The energy values are reported with respect to the ground state energy for the configuration 2p6 (E0 = -3.12289e+4 eV) which appears as 0.0 e+0 in the table. Each level is assigned an index number which is used in the transition table to refer to its energy level value.
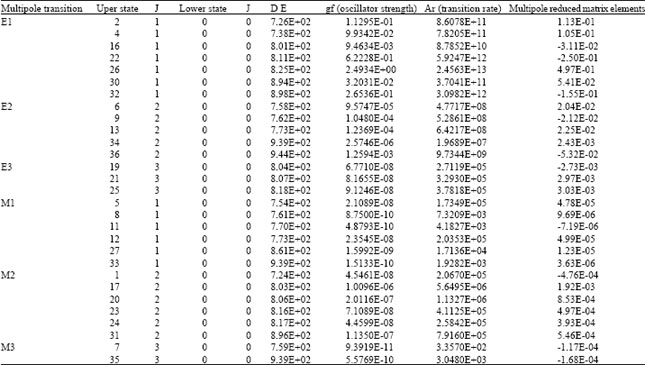
Transition rates of the optically allowed and forbidden (multipole) transitions from the excited states to the ground state are calculated. Each rank of multipole transition is calculated by a single run. Electric dipole (E1), electric quadrapole (E2) and (E3). magnetic dipole (M1), magnetic quadrapole (M2) and (M3) transition rates are listed in Table 2. Upper and lower states are defined by the numbers of their level indices. The transition rates of the allowed transitions (E1) are found to be the strongest. E1 transition rates less than ~8E+10 were ignored in the table. E2 transitions exhibit the strongest forbidden transitions. However, forbidden transitions are found to be weak in general as compared to the E1 transitions.
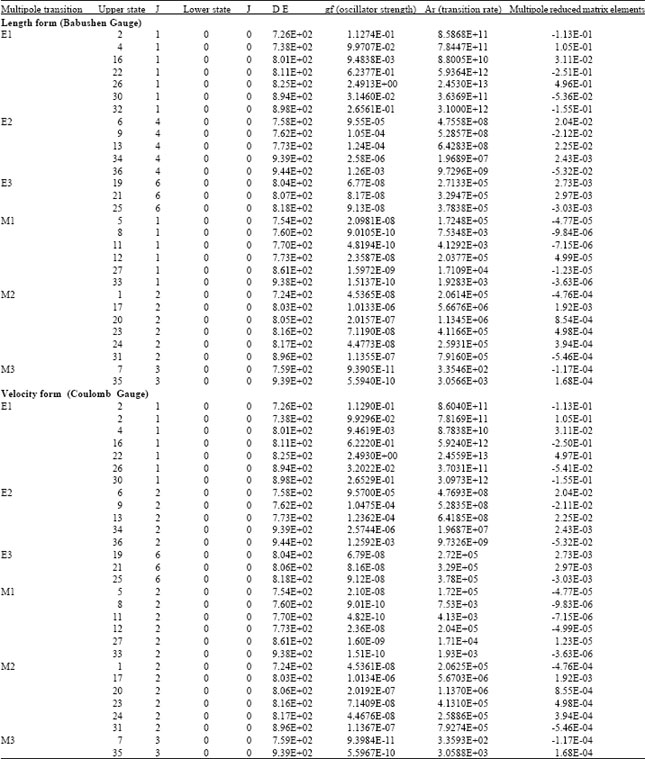
Multi reference many body perturbation theory: Table 3 presents the energy levels of Ne-like Fe ion as determined by the MR-MBPT option in HTAC. The length and velocity forms of the transition rates are considered (the produced data in the two forms were very close to each others).
By comparing data obtained through the RCIM and MR-MBPT options (Table 2, 3) we learn that values have differences in the range (10-5-10-1)%.
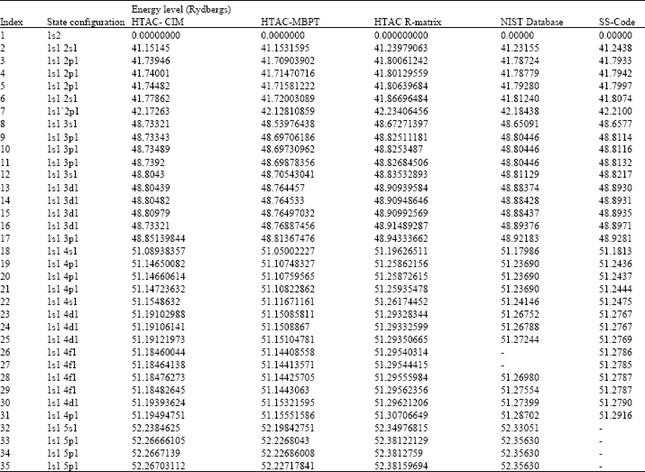
Validating HTAC produced atomic data: Here, produced atomic data for OVII ion (energy levels, radiative rates and oscillator strength) using HTAC code are presenting and compared with values compiled by the National Institute of Standards and Technology (NIST) produced by and the comprehensive theoretical data produced by Delahaye and Pradhan (2002) who adopted the Super Structure (SS) program of Eissner et al. (1974).
| Table 1: | Calculated energy levels for the Ne-like Fe by HTAC |
 | |
| Table 2: | Allowed and forbidden transitions as calculated by the CIM option in HTAC |
 | |
| Table 3: | Allowed and forbidden transition rates generated through the MR-MBPT option in HTAC |
 | |
Calculated Energy levels and radiative rates are presented in Table 4. As can be learned from Table 4, energy levels obtained using HTAC’s three methods are in good agreement (confidence ~99%) with NIST values and SS-code theoretical values.
| Table 4: | Energy levels for OVII ion produced using HTAC code validated against published experimental and theoretical data |
 | |
CONCLUSIONS
HTAC is a new atomic calculation code that has been developed to perform atomic structure and transition rate calculations for allowed and forbidden transitions in a simplified way using three advanced atomic calculation methods; the fully relativistic configuration interaction method, the multi reference many body perturbation theory and the R-Matrix method. HTAC is featured with a user friendly interface that enables researchers to perform atomic calculations through Windows instead of going through the hassles of other command-based codes. It also enables researchers to view and plot produced atomic data using options provided in the code. HTAC has a built in data base for all elements in the periodic table. It also allows the user to construct own input data files. Atomic structure program generates four different output files with four different extensions based on the computational method used to generate them. Produced data come up in four different tables; the energy levels table, the radiative transition rates table wave function table and the basis and mixing coefficients tables. HTAC atomic structure program has been used successfully to produce accurate atomic data for Ne-like Fe and OVII ions.
REFERENCES
- Busquet, M., A. Bar-Shalom, M. Klapisch and J. Oreg, 2006. An improved version of the HULLAC code. J. Phys. IV France, 133: 973-975.
CrossRef - Chernysheva, L.V. and V.L. Yakontov, 1999. Two-program package to calculate the ground and excited state wave functions in the Hartree-Fock-Dirac approximation. Comput. Phys. Commun., 119: 232-255.
CrossRef - Delahaye, F. and A. Pradhan, 2002. Electron impact excitation of helium-like oxygen up to n = 4 levels including radiation damping. J. Phys. B: At. Mol. Opt. Phys., 35: 3377-3377.
CrossRef - Zanna, D.G., M.C. Chidichimo and H.E. Mason, 2005. Benchmarking atomic data for astrophysics: FeXXIII. Astron. Astrophys., 432: 1137-1150.
Direct Link - Eissner, W., M. Jones and H. Nussbaumer, 1974. Techniques for the calculation of atomic structures and radiative data including relativistic corrections. Comp. Phys. Comm., 8: 270-306.
CrossRef - Ekuma, C.E., 2007. Rigorous solution of quantum scattering theory. Res. J. Phys., 1: 19-26.
CrossRefDirect Link - Gaigalas, G., Z. Rudzikas and C.F. Ficher, 1997. An efficient approach for spin - angular integrations in atomic structure calculations. J. Phys. B: Atomic Mol. Optical Phys., 30: 3747-3747.
CrossRef - Gupta, G.P. and A.Z. Msezane, 2009. Large-scale CIV3 calculations of fine-structure energy levels and radiative rates in Al-like copper. Can. J. Phys., 87: 895-907.
CrossRef - Hibbert, A., 1975. CIV3-A general program to calculate configuration interaction wave functions and electric-dipole oscillator strengths. Comp. Phys. Commun., 9: 141-172.
CrossRef - Ishikawa, Y., J.A. Santana and E. Trabert, 2010. Relativistic multireference many-body perturbation theory for open-shell ions with multiple valence shell electrons: The transition rates and lifetimes of the excited levels in chlorinelike Fe X. J. Phys. B: Atomic Mol. Optical Phys., 43: 074022-074022.
CrossRef - Safronova, M.S., W.R. Johnson and U.I. Safronova 1996. Relativistic many-body calculations of the energies of n=2 states for the beryllium-like isoelectronic sequence. Phys. Rev. A., 53: 4036-4053.
CrossRef - Safronova, U.I., A.S. Safronova, S.M. Hamasha and P. Beiersdorfer, 2006. Relativistic many-body calculations of multipole (E1, M1, E2, M2, E3 and M3) transition wavelengths and rates between 3l-14l' excited and ground states in nickel-like ions. Atomic Data Nucl. Data Tables, 92: 47-104.
CrossRef - Zhang, H.L., D.H. Sampson and A.K. Mohanty, 1989. Fully relativistic and quasirelativistic distorted-wave methods for calculating collision strengths for highly charged ions. Phys. Rev. A, 40: 616-632.
PubMedDirect Link - Vilkas, M.J, Y. Ishikawa and K. Koc, 1998. Second-order multiconfigurational Dirac-Fock calculations on boron-like ions. Int. J. Quantum Chem., 70: 813-823.
CrossRef - Vilkas, M.J., Y. Ishikawa and E. Trabert, 2007. Relativistic multireference many-body perturbation theory calculations on Au64+-Au69+ ions. Eur. Phys. J. D. 41: 77-93.
CrossRef