
Iiro Pulkkinen
Department of Biotechnology and Chemical Technology, Helsinki University of Technology, P.O. Box 6100, FI-02015, Helsinki, Finland
Juha Fiskari
Oy Metsä-Botnia Ab, Joutseno Mill, FI-54120, Finland
Ville Alopaeus
Department of Biotechnology and Chemical Technology, Helsinki University of Technology, P.O. Box 6100, FI-02015, Helsinki, Finland
Journal of Applied Sciences
Year: 2009 | Volume: 9 | Issue: 22 | Page No.: 3991-3998







ABSTRACT
The objective of this study was to examine the crystallinity of the cellulose of seven fully bleached pulp samples by using solid-state cross-polarization/magic angle spinning (CP/MAS) 13C Nuclear Magnetic Resonance (NMR) spectra in combination with line-fitting analysis and how it affected the hygroexpansional properties of handsheets. Differential scanning calorimetry was used to measure the pore size distribution of the samples and to characterize the water inside the fiber wall. Combined with the CP/MAS 13C NMR analysis, mechanisms behind the moisture absorbing capacities of various hardwoods were studied. Based on the results, the hygroexpansional behavior of the handsheets and the Crystallinity Index of the cellulose in the samples were not inter-correlated. The fiber wall thickness measured with a FiberLab® fiber analyzer had a moderate correlation with the amount of crystalline cellulose and xylan of the samples, implying that the crystalline and the amorphous regions were evenly distributed in the fiber wall.
PDF Abstract XML References Citation
How to cite this article
Iiro Pulkkinen, Juha Fiskari and Ville Alopaeus, 2009. CPMAS 13C NMR Analysis of Fully Bleached Eucalypt Pulp Samples: Links to Handsheet Hygroexpansivity and Strength Properties. Journal of Applied Sciences, 9: 3991-3998.
DOI: 10.3923/jas.2009.3991.3998
URL: https://scialert.net/abstract/?doi=jas.2009.3991.3998
DOI: 10.3923/jas.2009.3991.3998
URL: https://scialert.net/abstract/?doi=jas.2009.3991.3998
INTRODUCTION
Solid state CP/MAS 13C NMR spectroscopy is a versatile tool for morphology studies of cellulosic materials (Atalla and Hart, 1999; Hult et al., 2001, 2003). The supra-molecular structures of cellulose were investigated by using CP/MAS 13C NMR spectra along with non-linear spectral fitting, which analyzes the crystalline allomorph and disordered domains (Larsson et al., 1995, 1997; Wickholm et al., 1998; Hult et al., 2003). The line-shape spectral fitting analysis of solid state spectra allows a very detailed comparison and characterization of cellulose supra-molecular structures and has been extensively used to investigate the structural characteristics of cellulose and its derivatives. The relative amounts of different cellulose forms in complex cellulosic materials can be determined by using spectral fitting analysis of the C4-region of CP/MAS 13C NMR spectra (Fig. 1). The ratio of crystalline and amorphous cellulose can be characterized with a crystallinity index (CrI) (Newman and Hemmingson, 1990; Liitiä et al., 2003).
The microfibrils in bleached chemical pulp fibers consist of two regions of cellulose: crystalline and amorphous (Bertran and Dale, 1986). There is also a region between these two forms of cellulose, normally referred as para-crystalline cellulose (Larson et al., 1997). The highly ordered cellulose fibrils are barely accessible to water molecules. The amorphous regions consisting of less-ordered cellulose molecules and hemicelluloses absorb water. In cell walls, only 60-70 % of cellulose chains are in crystalline form (Alince, 2002). The rest are considered disordered (amorphous) and fully accessible to water molecules. A trace amount of adsorbed water on the surface of crystalline region is almost negligible, the amount of absorbed water in fibers, also called the bound water, almost totally relies on the accessible sorption sites of amorphous regions.
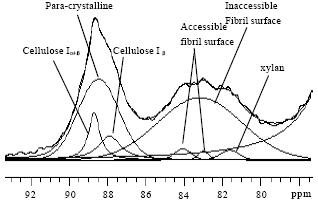 | |
| Fig. 1: | Spectral fitting for the C-4 region of the CP/MAS 13C NMR spectrum of a birch kraft pulp sample |
Determination of the size of amorphous region of fibers can be conducted by Differential Scanning Calorimetry (DSC), which is commonly used for determination of bound water in fibers (Maloney et al., 1998; Salmén and Berthold, 1997). In this study, DSC was used to characterize the pore structure for different hardwood fibers. Focus was also placed on the effect of cellulose crystallinity on the pore size distribution.
Hygroexpansion is a paper property of great interest in many applications, for instance in printing operations, but also in relationship to mechano-sorptive creep. Paper expands or shrinks as the relative humidity in the local environment changes. When hygroexpansivity is plotted against relative humidity, paper generally shows complex hygroexpansion behavior, dependent on its humidity history before and during testing (Uesaka, 2002; Salmén, 1993). Sheet dimensional changes occur in concert with the changes in fiber dimensions. At the hydrogen-bonded fiber crossing areas, the more stable direction of a fiber is able to restrict change in the less stable direction of another, so that intra-planar dimensional changes are far smaller than those in the z-direction.
The hygroexpansivity is highly influenced by restrictions affecting the orientation of both the cellulose and the hemicellulose chains (Salmén et al., 1987a, b; Uesaka, 2002). The dried-in stresses in hemicelluloses can be released by increasing the relative humidity to about 50-60%, thus increasing the hygroexpansivity of paper if it has been dried under restraint to very low humidities. As hemicelluloses and the amorphous cellulose are responsible for the water uptake of pulp fibers (Salmén and Berthold, 1997), NMR studies provide information about the influence of the crystallinity degree and the xylan content in the fiber wall on hygroexpansivity and strength properties.
The purpose of this investigation was to examine how various hardwood samples differ in terms of their degree of cellulose crystallinity and their pore size. With the information obtained, the characteristics of the hygroexpansional behavior of handsheets made of bleached hardwood kraft pulp samples in relation to cellulose crystallinity can be studied. Statistical analysis was used to find links between the NMR data and the strength and structural properties of the handsheets.
MATERIALS AND METHODS
Pulp samples and carbohydrate testing: Bleached industrial hardwood kraft pulps (Eucalyptus grandis E. dunnii, Uruguay; Betula pendula/B. pubescens, Finland; Acacia magnium, Indonesia; E. globulus, Portugal; E. urograndis/E. grandis, South Africa; E. grandis/E. saligna, Brazil; E.urograndis, Brazil) were obtained from pulp mills as dry sheets. Laboratory handsheets with a grammage of approximately 60 g m-2 were prepared in a conventional sheet former according to ISO5269-1:1998. Anisotropic sheets were made with a dynamic sheet former at KCL Research Center. The wire speed was set to 1000 m min-1. The jet speed was set to 920 m min-1 (1.6 bar). The feed pulp concentration was 4 g L-1. The grammage of anisotropic sheets was 60 g m-2. Tensile properties were measured according to EN ISO 1924 and tensile stiffness was measured according to SCAN-P 67:93. The handsheets were prepared and tested at KCL research center (Espoo, Finland).
The carbohydrate composition of the pulps was determined using the following method: utilizing sulfuric acid hydrolysis at 120°C to degrade the carbohydrates into monosugars, based on Tappi standard T222-om0. The monosugars were then analyzed by ion chromatography (Metrohm 817 Bioscan system). The carbohydrate measurements were conducted in Botnia mill at Kemi.
The FiberLab® measurement apparatus consists of an analyzer and a s ample unit. Fiber images are captured by two CCD cameras. The direct results are the fiber length, fiber width and fiber wall thickness. The calculated values are the curl index, coarseness, cross-sectional area and volume index. Procedures of fiber and image processing to obtain fiber properties have been described in detail elsewhere (Anonymous, 2006). Kajaani FiberLab® data for the hardwoods was recorded at the Botnia mill in Kemi. This study was conducted in Espoo, Finland, from 2nd of January to 5th of June 2009.
NMR measurements: The solid-state CP/MAS 13C NMR experiments (Atalla and Hart, 1984) were performed on a Bruker Avance-400 spectrometer operating at frequencies of 100.59 MHz for 13C. All the experiments were carried out at ambient temperature using a Bruker 4-mm MAS probe. The pulp samples (~45% moisture) were packed in a 4 mm ZrO rotor. CP/MAS 13C NMR data was acquired with 8000 scans accumulated per sample. The samples were analyzed at Georgia Institute of Technology, Atlanta, US.
Pore measurements: The thermoporosimetry measurements were carried out using a Mettler DSC 821. In the technique used in this study, the energy that is absorbed when water in frozen pulp fibers is melted in a stepwise manner approaching 0°C is measured. The melting energy is assumed to be directly proportional to the amount of melted water. This technique is based on the fact that water contained within pores sufficiently small or inside a swollen hemicellulose gel is at an elevated pressure and therefore has a depressed melting temperature. The details of the measurements have been described by Maloney and Paulapuro (1999). The pore size measurements were conducted at Helsinki University of Technology.
Measurement of hygroexpansion: Hygroexpansion was measured on test pieces subjected to a relative humidity of 66% RH prior to testing. A non-standardized apparatus based on laser measurement of the dimensional change in the sample length was used to measure the hygroexpansion of paper strips. The measurements were performed at STFI Packforsk, with an instrument similar to a previous version described by Salmén (1993). It consists of 30 rigid clamps and 30 freely movable clamps, with 100-mm gaps between the clamps, in which 30 paper strips are placed independently in a horizontal position. A weight is placed on the strips to eliminate any effects ofbuckling on length registration. These clamps were placed in a chamber with regulated humidity. The relative humidity was changed from 50±2%RH to 22±3%RH to 33±2%RH to 66±2%RH and the length changes between 33±2%RH and 66±2%RH were measured.
The hygroexpansion coefficient (β) was determined from the gradient of the linear part of the curves in the low-moisture range and is expressed as the hygroexpansional strain divided by the moisture change of the sample (Nanri and Uesaka, 1993).
| (1) |
Data analysis of the results: Table 1 shows the results of the correlation analysis (Milton and Arnold, 1995) performed between the measured and calculated pulp fiber properties, the handsheet properties of un-refined fibers and the NMR data. Since, the number of samples was only seven, the critical value for making the results statistically significant for a two-tailed test (<5%) was 0.75. Table 2 shows the correlations between the NMR data and the hygroexpansivity measurements.
| Table 1: | Correlations of NMR data, handsheet strength properties and fiber dimensions (2-tailed test, statistically significant (p<0.05). Level of significance > 0.74 (7 samples) |
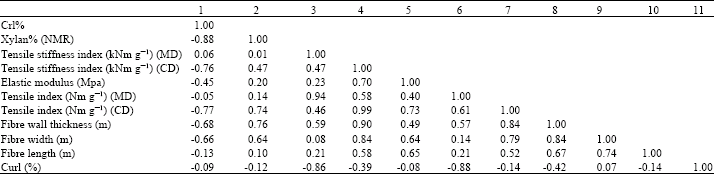 | |
| A correlation analysis on the crystallinity index and xylan content (relative amount in percent), strength properties of dynamic handsheets in machine and cross direction and fibre characteristics (fibre length, widgth, wall thickness and fibre curl). 1: Crl%, 2: Xylan% (NMR), 3: Tensile stiffness index (kNm g-1) (MD), 4: Tensile stiffness index (kNm g-1) (CD), 5: Elastic modulus (Mpa). 6: Tensile index (Nm g-1) (MD), 7: Tensile index (Nm g-1), (CD), 8: Fibre wall thickness (m), 9: Fibre width (m), 10: Fibre length (m), 11: Curl (%) | |
| Table 2: | Correlations of NMR data and the hygroexpansivity measurements (2-tailed test, statistically significant (p<0.05). Level of significance>0.74 (7 samples) |
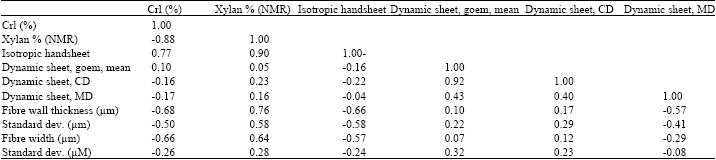 | |
| A correlation analysis on the crystallinity index and xylan (measured with NMR in relative amount in percent), the hygroexpansional properties of isotropic and dynamic handsheets and the mean and standard deviation of fiber width and fiber wall thickness. The hygroexpansitivities obtained from Pulkkinen et al. (2009b) | |
| Table 3: | The line-fitting results of the C-4 region of CP/MAS 13C NMR spectrum of pulp sample (values are relative intensity as a percentage, %) |
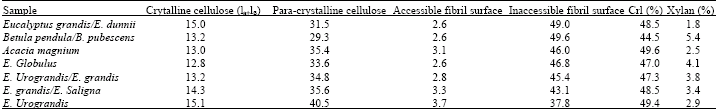 | |
| Values taken from Pulkkinen et al.(2009b) | |
RESULTS
The crystallinity index (CrI) has been considered an important factor for monitoring the structural features of the cellulosic substrate. The calculated crystallinity index (CrI) values are presented in Table 3 along with the rest of the results of the spectral fitting. CrI was calculated as a percentage of the sum of crystalline and para-crystalline cellulose fraction in the C-4 signal cluster (Lennholm et al., 1994). The correlation between the values determined by ion chromatography and those determined by CP/MAS 13C NMR was good (Fig. 2), implying that there is a certain fraction of xylan that is intimately associated with the cellulose, as has been suggested for bleached birch kraft pulp (Liitiä et al., 2003). The correlation between the results of carbohydrate analysis and the relative amount of xylan observed from the spectral analysis also gives us confidence that xylan amounts between samples of this study are comparable. Recent evidence of the association between xylan and cellulose based on dynamic FT-IR has also been presented by Dammström et al. (2009).
Based on Table 1 and 2, the crystallinity index (CrI) and xylan content of the samples had good correlations with tensile strength properties in the cross direction and moderate correlations with the fiber wall thickness index and fiber width. With an increasing cellulose crystallinity, tensile strength in the cross-direction decreased, probably due to the lower amount of xylan in the fiber wall (Table 1). With higher xylan contents the strength of the handsheets increased. This kind of behavior with regard to strength properties has been confirmed earlier (Annergren et al., 1963; Kettunen et al., 1982; Kibblewhite and Bawden, 1989), as samples with a higher amount of crystalline cellulose produce sheets with lower tensile strength.
The only significant correlation observed for the hygroexpansional data was between the hygroexpansivity of the handsheets and the xylan content of the fiber wall. The negative correlation can be at least partly explained by increasing fiber wall thickness value as the xylan content increases. More coarse fibers decrease the hygroexpansivity coefficient of the handsheets (Pulkkinen et al., 2009a).
There seems to be no apparent correlation between the measured hygroexpansion coefficients and the cellulose crystallinity index.
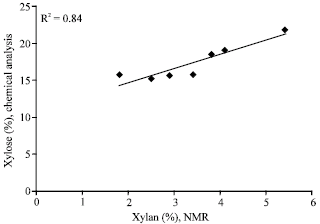 | |
| Fig. 2: | The correlation between xylan measured chromatographically and xylan measured with NMR |
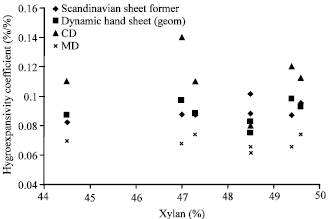 | |
| Fig. 3: | Hygroexpansivity of isotropic handsheets (diamonds) and anisotropic (dynamic mean value) handsheets; in CD (triangles), in MD (crosses) and the geometric mean (squares) for laser measurement apparatus (Pulkkinen et al., 2009b) |
Only a weak correlation was observed for conventional handsheets (Table 2). The observed values for hygroexpansion of conventional and dynamic handsheets were extracted from our previous publication (Pulkkinen et al., 2009b) and are shown here as a function of crystallinity index in Fig. 3.
The water absorption capacity of pulp fibers due to the amount of amorphous regions of the fibers wasindirectly detected with the Differential Scanning Calorimetry (DSC). The results are shown in Table 4 forthe pulp samples studied. The amount of non-freezing water and bound water obtained with the DSC should be quite similar for never-dried and dried samples (Salmén and Berthold, 1997; Maloney and Paulapuro, 1999). Therefore, the differences in drying of the pulp samples were not assumed to cause any differences in the measurements with the DSC (Salmén, 1993; Maloney and Paulapuro, 1999).
Since, the pulps were obtained from different sources, the fact that the amount of lignin and hemicelluloses varies between samples has to be taken into account. This will affect the amount of bound water measured with the DSC (Salmén and Berthold, 1997) as pore sizes develop into larger ones when the removal of lignin and hemicelluloses progresses further. As shown in Fig. 4a and b, the mean pore size measured with the DSC is a function of both the crystallinity index and the amount of xylan measured with NMR. Therefore, it seems that fibers with low xylan content had a slightly lower mean pore size independent of the amount of water contained in the amorphous regions of the fiber wall (Table 4). This is consistent with the observations by Duchesne et al. (2001). They observed that the fibril aggregates (macrofibrils) with high hemicellulose content were well organized with a porous structure. Fibril aggregates with low hemicellulose content had a smoother, flatter and more compact surface structure.
No correlation between the non-freezing water content and the crystallinity of the cellulose was observed. The amount of non-freezing water was approximately constant for all samples (~0.3 g/g). The fiber wall thickness measured with a FiberLab® analyzer and the crystallinity of the cellulose had a moderate correlation with the crystallinity index (Fig. 5).
The interpretation of the xylan from the spectral data is not straightforward and should be handled with caution (Liitiä et al., 2003). Therefore, the observed weak correlation between the fiber wall thickness and the xylan content (Table 1) may be due to the mild affinity between the cellulose and the hemicelluloses.
As a fiber analyzer can only detect the birefringent part of the fibers, i.e., cellulose (Piirainen, 1985; Olsson et al., 1995), the fiber wall thickness value may be partly influenced by the fiber detection procedure (Jordan and O’Neill, 1994). Keeping in mind the principles of fiber wall detection with an analyzer, the most important observation is the correlation between the fiber wall thickness index and the crystallinity of the cellulose.
The increased hydrogen bonding, where the fibrils can form tighter aggregates when the amount of xylan decreases, seems to affect the strength properties of thefiber network. If the effect of the transverse dimensions of the fibers on network strength is based on the open micropore structure present in un-dried fibers, after drying the transverse dimensions lose their impact on the strength properties unless refining is applied.
The hygroexpansion coefficient only covers the low range of moistures of the samples (Nanri and Uesaka, 1993). It has been previously observed by Pulkkinen et al. (2009a) that samples that indicate a low hygroexpansivity coefficient often undergo major changes in sheet dimensions across the whole humidity range.
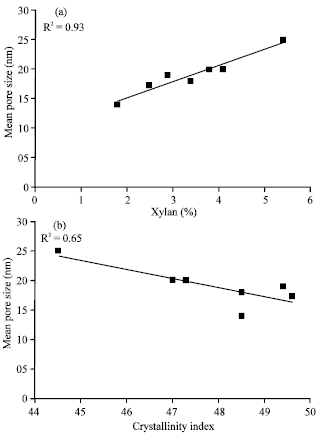 | |
| Fig. 4: | The mean pore size as a function of the (a) Crystallinity Index (CrI)(right) and as a function of (b) xylan measured with NMR (left) |
| Table 4: | Results of the DSC analysis. NFW = non-freezing bound water (mL g-1) |
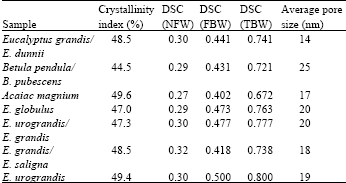 | |
| FBW: Free bound warter (mL g-1), TBW: Total bound water (mL g-1) | |
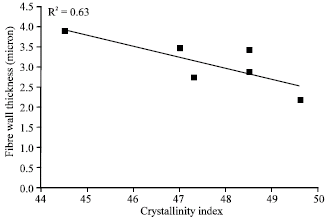 | |
| Fig. 5: | Fiber wall thickness as a function of the crystallinity index |
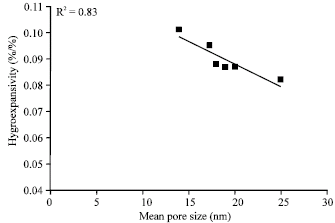 | |
| Fig. 6: | Hygroexpansivity of isotropic handsheets as a function of mean pore size measured with a DSC |
Low hygroexpansivity is believed to be associated with a high fiber wall thickness index (Pulkkinen et al., 2009a; Nanko and Wu, 1995).
The low amount of total water absorbed by the sample of A. magnium was probably affected by the low amount of xylan in the fiber wall. The amount of bound water in the sample was by far the lowest among those studied (Table 4). Also, the small average pore size probably affected the moisture absorbance of the fibers, enabling them to absorb water at the fastest rate, resulting in a high hygroexpansion coefficient. Further evidence is given by Fig. 6, which shows the hygroexpansivity of an isotropic handsheet plotted against the average pore size.
The findings of this study give us reason to speculate that the differences seen between the strength properties of the unrefined samples are dependent on the crystallinity degree of the cellulose and on the transverse dimensions of the fibers. The hygroexpansivity of isotropic handsheets were seen to be dependent on theaverage pore size of the fibers. The amount of amorphous regions and xylan was also a determining factor in isotropic hygroexpansion.
DISCUSSION
The algorithm used in this study was not able to include Lorentzian lines for modeling the signals originating from the highly crystalline domain in C(4) region (Larsson and Westlund, 2005) and only Gaussian lineshapes were used. In this study, the Iβ form of crystalline cellulose (Atalla and Hart, 1984; Larsson and Westlund, 2005) was not observed for the hardwood samples measured. However, it is possible that this was a feature of the measurement accuracy of the spectral analysis applied. Because of the previous facts it is possible that the values for the Crystallinity Index or the relative amount of xylan were not as accurate as those obtained with combined use of Lorenzian and Gaussian lines. However, all studied spectra were treated in the same manner, so obtained results are comparable within this study.
The moderate correlation between fiber wall thickness, fiber width and CrI is an interesting and new finding. Based on this study, the crystallinity of the fiber wall decreases (and the xylan content increases), as the fiber wall thickness increases. One probable reason is the birefringence, i.e., double refraction, of particles containing cellulose (Piirainen, 1985; Olsson et al., 1995). This would mean that the hemicelluloses and amorphous regions of fibers would be undetectable in a fiber analyzer. This may also hinder the observation of the linkage between xylan content and hygroexpansivity. This contradicts with the positive correlation between xylan and fiber wall thickness index observed in this study. This can be partly due to the higher swelling ability of the outer surface of the fiber due to the higher xylan content (Dahlman et al., 2003). This can promote more easily detectable outer surface for a scanned fiber flowing through a measurement capillary, as cellulose fibrils and xylan are in close association with each other.
The detection of cellulose-xylan-matrix with the optical analyzers needs to be studied in more detail to shed some light over the influence of the location of the xylan on the detection process. Dahlman et al. (2003) have observed that the outer layer (S1) of hardwood pulp fibers have a higher relative content of xylan than the corresponding inner layer (S2+S3). However, regarding the samples of this study, it is safe to assume that the S1 layer has been removed during the pulp manufacturing process. The correlation between the xylan contents and the hygroexpansivity of conventional handsheets and the absence of correlation between the xylan content and oriented sheet seem to imply that obtaining similar orientation for all the dynamic sheets was unsuccessful. However, in our previous study (Pulkkinen et al., 2009a), the effect of anisotropy, measured as the MD/CD ratio of tensile strengths, was observed to be similar for allsamples. Due to the fact that fibrils are always laid at an angle to the fiber axis, the orientation of the hemicellulose chains may have more influence on the cross-fiber dimensions, thus affecting the hygroexpansivity of the paper.
CONCLUSIONS
The objective of this study was to investigate how hardwood fibers that differ in their ultrastructure and dimensions perform mechanically and what the effect of ultrastructure is on the hygroexpansivity of paper. Based on these results, the hygroexpansional behavior of the handsheets and the crystallinity index of the cellulose in the samples were not inter-correlated. The fiber wall thickness had a moderate correlation with the amount of crystalline cellulose in the samples, implying that the crystalline and the amorphous cellulose were evenly distributed in the fiber wall. The observed correlation between fiber wall thickness and xylan content could be due to the mild affinity between the cellulose and hemicelluloses. The water-absorbing capability of the fibers measured with a DSC did not give indications of the hygroexpansional behavior of the fiber network.
The findings of this study led us to speculate that the differences seen between the strength properties of the unrefined samples used in this study were mainly dependent on the crystallinity degree of the cellulose and the transverse dimensions of the fibers. The dimensional stability of the dynamic sheet samples was not affected by the crystallinity of the cellulose. The distinction between the effects of xylan in fibers and fiber surfaces and fiber wall thickness could be investigated in a separate study using fibers with similar dimensions and varying amounts of xylan.
ACKNOWLEDGMENTS
Sirkka-Liisa Maunu for her valuable comments during the preparation of this manuscript. Professor Arthur Ragauskas and Dr. Yunqiao Pu for providing the results of NMR spectroscopy.
REFERENCES
- Atalla, R.H. and D.L.V. Hart, 1999. The role of solid state 13 C NMR spectroscopy in studies of the nature of native celluloses. Solid State Nucl. Magnetic Resonance, 15: 1-19.
PubMed - Dahlman, O., A. Jacobs and J. Sjoberg, 2003. Molecular properties of hemicelluloses located in the surface and inner layers of hardwood and softwood pulps. Cellulose, 36: 325-325.
Direct Link - Hult, E.L., P.T. Larsson and T. Iversen, 2001. Cellulose fibril aggregation an inherent property of kraft pulps. Polymer, 42: 3309-3314.
CrossRef - Hult, E.L., T. Iversen and J. Sugiyama, 2003. Characterization of the supermolecular structure of cellulose in wood pulp fibers. Cellulose, 10: 103-110.
CrossRef - Larsson, P.T., U. Westermark and T. Iversen, 1995. Determination of the cellulose Iα allomorph content in a tunicate cellulose by CP/MAS 13 C-NMR spectroscopy. Carbohydr. Res., 278: 339-343.
CrossRef - Larsson, P.T., K. Wickholm and T. Iversen, 1997. A CP/MAS13 C NMR investigation of molecular ordering in celluloses. Carbohydr. Res., 302: 19-25.
CrossRef - Liiti�, T., S.L. Maunu, B. Hortling, T. Tamminen, O. Pekkala and A. Varhimo, 2003. Cellulose crystallinity and ordering of hemicelluloses in pine and birch pulps as revealed by solid-state NMR spectroscopic methods. Cellulose, 10: 307-316.
Direct Link - Atalla, R.H. and D.L. Vanderhart, 1984. Native cellulose. Composite of two distinct crystalline form. Science, 223: 283-285.
CrossRef