
Hamzeh S.M. Al-Omari
Department of Chemistry, College of Science, Mutah University,
P.O. Box 7, Karak 61710, Jordan
Journal of Applied Sciences
Year: 2008 | Volume: 8 | Issue: 15 | Page No.: 2659-2668







ABSTRACT
Amino-oxo cytosine and amino-hydroxy cytosine tautomeric system was studied by employing MNDO semi-empirical method of calculations. The amino-hydroxy structure was found to be energetically favorable over amino-oxo form of cytosine as indicated by the calculated heat of formation and Gibbs free energy. The substitution of F, OH, CN, NH2, BH2 and CH3 at carbon 6-position was studied. All the substituents are found to affect the geometrical parameters of the substituted structures. The equilibria between the substituted amino-oxo and amino-hydroxy cytosines were studied by the Gibbs free energy change and by isodesmic reactions. The calculations were performed at 25C. It is found that all substituents push the equilibria in the forward direction compared to the unsubstituted tautomeric pairs except in the case of BH2 substituent where the equilibrium becomes slightly less favourable in the forward direction.
PDF Abstract XML References Citation
How to cite this article
Hamzeh S.M. Al-Omari, 2008. Effect of Different Substituents on the Amino-Oxo/Amino-Hydroxy Cytosine Tautomeric System. Journal of Applied Sciences, 8: 2659-2668.
DOI: 10.3923/jas.2008.2659.2668
URL: https://scialert.net/abstract/?doi=jas.2008.2659.2668
DOI: 10.3923/jas.2008.2659.2668
URL: https://scialert.net/abstract/?doi=jas.2008.2659.2668
INTRODUCTION
The interest in this study has stemmed from the importance of cytosine as a fundamental part of the DNA structure. In the process of DNA duplication cytosine pairs guanine with strong hydrogen bonding from three different sites, this hydrogen bonding with guanine (Lewin, 1983) will not be possible when the cytosine tautomer occurs for some reason and therefore leads to spontaneous mutation (Topal and Fresco, 1976; Blackbon and Gait, 1996).
Cytosine molecule may exist in various tautomeric forms which differ from each other by the position of the acidic hydrogen atoms in the molecule, where they may bind to either ring nitrogen or oxygen atoms or both (Wolken et al., 2007; Yao et al., 2007; Forgarasi and Szalay, 2002; Fazaeli et al., 2002; Civcir, 2000).
All of the previous studies were concentrated on the stability of the possible cytosine tautomers theoretically (Wolken et al., 2007; Zhao et al., 2006; Chung et al., 2005; Podolyan et al., 2003; Piacenza and Grimme, 2003; Forgarasi and Szalay, 2002; Sambrano et al., 2000; Fogarasi, 1997; Estrin et al., 1994) and experimentally (Fazaeli et al., 2002; Brown et al., 1989; Orozco et al., 1998; Nowak et al., 1989; Gould et al., 1992; Jaworski et al., 1990) in gas and in aqueous phases.
Even though lots of work have been done on the cytosine tautomeric system, but only a few of them have reported the effect of substituents on the stability of cytosine and its tautomers. One study was found in which the CH3, Cl, propenyl and Br substituents were studied at position 5 in the gas and aqueous phases by ab initio method for the amino/imino tautomerism (Rueda et al., 2001), another one was the effect of substitution of CH3 group at position 5 in gas and aqueous phases by DFT method (Sambrano et al., 2001).
The aim of this research is to study the effect of substituents, at position 6, on the amino-oxo/amino-hydroxy cytosine tautomeric system, which haven`t been studied before. The effect of substituents is shown through the free energy calculations of the different tautomeric systems supported by the results obtained from the isodesmic reactions.
Isodesmic reactions were first introduced by Benson (1965) and defined as reactions in which there is retention of the number of bonds of a given formal type, i.e., constant number of bonds of a given type between the reactants and products (Hehre et al., 1970), also these reactions are named as bond separation reactions. Due to the constant number and type of bonds, the isodesmic reaction energies can be calculated with high accuracy. Isodesmic reactions can be purely hypothetical and their energies can be used to estimate the stabilization effect of substituents on different compounds. This can be illustrated through the following explanation:
If X is used as substituent on compounds A and B, the isodesmic reaction, where A-X represents the X-substituted A and B-X represents the X-substituted B, is:
A-X + B → B-X + A isodesmic reaction
The enthalpy change for this reaction is given by:
Δhrxn = ΔHf(A) + ΔHf(B—X) —ΔHf(B) —ΔHf(A—X)
= [ΔHf(A) —ΔHf(A—X) ] — [ΔHf(B) —ΔHf(B—X)]
= First term-Second term
if the first term is larger than the second term this means that ΔHrxn is positive otherwise it is negative. A positive ΔHrxn would be represented in the following diagram where the difference between energy levels 1 and 2 (First term) is larger than that between levels 3 and 4 (second term). This difference in energy levels is the factor that determines weather ΔHrxn is positive or negative no matter where the energy levels themselves are. For example, the energy difference, between levels 1 and 2 represents the stabilization in A that occurs when it is reacted to form A—X. So, the diagram shows that the substituent X stabilizes compound A more than B.
| Δhf (A)_________(Level 1) | |||
| Δhf (B)_________(Level 3) | |||
| ΔHf (B—X)_________(Level 4) | |||
| Δhf (A—X)_________(Level 2) |
When the second term is larger, a negative ΔHrxn is produced and it can be said that A is destabilized by the substituent X relative to B, or B is more stabilized than A by the substituent X.
COMPUTATIONAL METHOD
Theoretical calculations were carried out by the use of MOPAC2007 program (Stewart, 2007), it is written in FORTRAN-90/95 language, it supports the MNDO (Dewar and Thiel, 1977) and other semi-empirical methods. The Self-consistent Field method used in MOPAC2007 is RHF. Baker`s Eigen Following procedure is used for geometry optimization. In this procedure, an approximate geometry is selected then the forces acting on the system are calculated, from which a new geometry can be calculated. The cycle is continued until reaching an equilibrium state, t this which, the energy is minimum and the net force acting on every atom in the system is very small or nearly zero. The geometry is called ground-state stationary geometry. Entropy and other thermodynamic quantities also can be calculated. The program has many other capabilities where vibrational spectra, force constants, isotopic substitution effect, transition states can be calculated.
RESULTS AND DISCUSSION
The parent cytosine and it`s tautomers
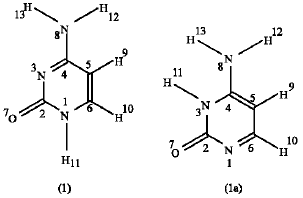
Cytosine (4-amino-1H-pyrimidine-2-one): The calculations in this work have been performed by the use of MNDO semiempirical method (Dewar and Thiel, 1977). No symmetry has been imposed throughout the calculations, i.e, all of the bond lengths and angles were allowed to optimize freely. The two possible oxo-amine forms depending on the position of the hydrogen atoms appear in scheme 1 below:
 |
| scheme 1 |
Structure 1 and 1a are termed as 4-amino-1H-pyrimidine-2-one and 4-amino-3H-pyrimidine-2-one respectively. In this work, it is found that the heat of formation of structure 1 is -48.134 kJ mol1 and that of structure 1a is -31.679 kJ mol1. These values indicate that structure 1 is more stable than structure 1a because it has lower heat of formation. The stability can be referred to the repulsion and attraction forces within these structures.
From these two structures it can be seen that the repulsion between (H10) and (H11) is absent in structure 1a and the repulsion between (H11) and (H13) is absent in structure 1. The other atoms are all similar in both structures. The repulsion between (H11) and (H13) in structure 1a is expected to be higher than that between (H10) and (H11) in structure 1. Both (H11) and (H13) in structure 1a are connected to highly electronegative atom (N) so, both of these hydrogens have relatively high positive charges. In structure 1, only (H11) is connected to a nitrogen atom, (H10) is connected to a carbon atom. The calculated charges are found to be 0.2009 and 0.1980 for (H11) and (H13) in structure 1a respectively and those of (H10) and (H11) in structure 1 are found to be 0.08211 and 0.2034, respectively. So, structure 1 is more stable than 1a and it will be considered in this study.
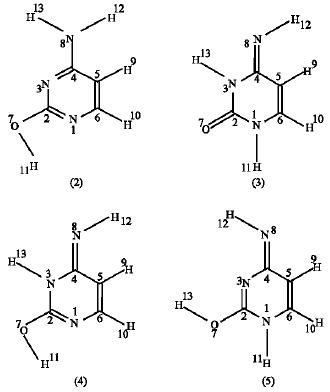
Cytosine tautomers: Besides the oxo-amino forms (1 and 1a) of cytosine, theoretically, other forms are also possible (scheme 2) depending on the possible positions of the acidic hydrogen atoms. Acidic hydrogens are those bonded either to nitrogen or to oxygen atoms in the structure.
 |
| scheme 2 |
Structure 2 is available in two rotamers, the other rotamer is with the hydroxyl group pointing up (syn), structure 2 is termed as anti. Structure 3 is also available in two rotamers and the other rotamer is with (H12) pointing to the left. Each of structures 4 and 5 is available in four different combinations of (H12) and hydroxyl groups orientations. The rotamers appear in scheme 2 are the most stable ones relative to their corresponding rotamers. The minimized repulsion between the atoms in these structures justifies their being of minimum energy as indicated from the charge distribution obtained. The order of stability of these structures is found to be 2>3>4>5. From these results, it can be said that the imino forms (3, 4, 5) are less stable than the amino forms (1 and 2), also the enol-imino forms are the least stable among all, this result is also found by some other workers when they used AM1 semiempirical method and ab initio calculations at different levels of theory (Colominas et al., 1996). When the amino forms are compared together, it is found that the amine-hydroxy (2) is more stable than amino-oxo (1), this means that structure (2) is the most stable among all of the forms of cytosine, this is referred to the aromaticity of this structure (Fogarasi, 1997) even though aromaticity is a relative term and has different measures (Cysewski, 2005). Experimentally, it is found that cytosine exists in the two amino forms (1 and 2) under Ar or N2 low temperature matrix and the imine tautomer (3) has not been identified (Estrin et al., 1994). An experimental study on generation of cytosine cations indicated that amino-hydroxy tautomer of cytosine is a major component in a cytosine sample in the gas phase (Yao et al., 2007). Other experimental work on the cytosine tautomers also shows that the amino-hydroxy tautomer (2) is available in substantial amount in the case of isolated molecules (Brown et al., 1989; Les and Adamowicz, 1989; Szczesniak, 1988). Piacenza and Grimme (2003) have used MP2, SCS-MP2, QCISD and QCISD(T) ab initio methods on their study on the DNA-base tautomers and they showed that the aromatic amino-hydroxy tautomer (2) is the most stable among all of the cytosine tautomers. They also performed the same calculations by the use of different DFT methods (HCTH407, PBE, BP86, B-LYP andBH-LYP) but they obtained the nonaromatic amino-oxo (2) as the most stable tautomer. Fazaeli et al. (2002) and Fogarasi (1997) in their theoretical work also obtained that hydroxyl-amino form (2) is the most stable structure when they used ab initio methods of calculations and the amino-oxo structure (1) is the most stable structure when they applied the DFT methods of calculations. The same result was obtained by other workers used B3LYP DFT calculations (Chung et al., 2005). A recent study performed by Wolken et al. (2007) applying B3LYP DFT and MP2(full) employing the highest level of theory basis set (6-31+G(d,p) also obtained that the anti-amino-hydroxy (1) as the most stable structure of cytosine. Therefore, the results obtained in this work are in good agreement with the experimental results and with those obtained from the ab initio calculations.
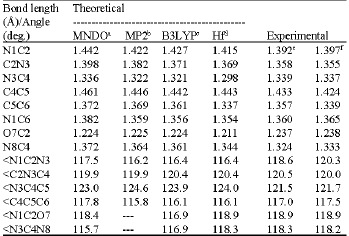
The calculated heat of formation of the amino-hydroxy tautomer (compound 2) is found to be -89.16 kJ mol1 and that of compound 1 is -48.13 kJ mol1 as mentioned earlier. The calculated entropies for compound 1 and 2 are 230.88 and 225.57 J mol1 K, respectively. Therefore, according to the equation, ΔG = ΔH—TΔS, (Attkins and de Paula, 2002) the calculated free energy change at 25C for the tautomerization reaction shown in scheme 3 below is -39.44 kJ mol1.
ΔG = -39.44 kJ mol1 | (1) |
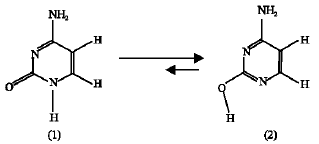 |
| scheme 3 |
The energy change (ΔH) for this reaction is -41.03 kJ mol1 and the entropy change is -5.31 J mol1 K, this value of the entropy change is negative which prefers the reactants (compound 1) but at 25C is not big enough in magnitude to overcome the large negative energy change which strongly prefers the products (compound 2), the net result is given by the largely negative free energy change (ΔG -39.44 kJ mol1) which prefers the product and so the equilibrium is shifted in the forward direction.
The geometrical parameters (bond lengths and bond angles) obtained in this work compared to those obtained by other workers, appear in (Table 1) for compound (1) and in (Table 3) for compound (2). No experimental data were found for the amino-hydroxy (2).
For the amino-oxo cytosine (1) (Table 1) it is noticed that N3C4 bond length obtained in this work (MNDO) is similar to the experimental (Voet and Rich, 1970; Clowney et al., 1999). O7C2 bond length is shorter compared to the experimental but similar to those obtained from MP2 (Fogarasi, 1997) and B3LYP (Sambrno et al., 2000). Shorter O7C2 bond length indicates preference of more carbonyl-like character (Wolken et al., 2007). The other bond lengths are higher than those obtained by MP2 and B3LYP methods (Table 1). The same observation can be said about the bond lengths in the case of the amino-hydroxy cytosine (2) (Table 3) where all of the bond lengths obtained by other theoretical methods are longer except the O7C2 bond which is shorter by MNDO method. Extensive discussion about the bond lengths and the geometry of cytosine can be found in many references (Fogarasi, 1997; Sponger and Hobza, 1994).
The dihedral angles obtained for the amino-oxo and amino-hydroxy compounds (1 and 2) are all either 0.00 or 180.00, this reflects planar geometry for these two structures. Planar structures were also obtained by Piacenza and Grimme (2003) when they used HCTH407, PBE, B3LYP, BH-LYP and HF methods of calculations. They obtained nonplanar structures (pyramidal NH2 group) with B-P and MP2 methods and showed that these differences in geometry had minor effect on the calculated energy and did not influence the order of stability of the cytosine tautomers, this is also found by other workers (Kobayashi, 1998; Chung et al., 2005; Sponger and Hobza, 1994).
| Table 1: | Selected bond lengths and bond angles of the amino-oxo cytosine (compound 1) obtained in this work compared to other theoretical and experimental results |
 | |
a: Present study; b: Fogarasi, 1997; c: Sambrano et al. (2000); d: Scanlan and Hillier (1984); e: Voet and Rich (1970); f: Clowney et al. (1999) | |
| Table 2: | Selected bond lengths and bond angles of the amino-hydroxy cytosine (compound 2) obtained in this work compared to other theoretical work |
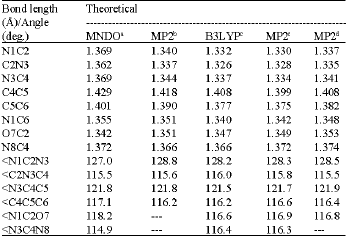 | |
a: Present study; b: Fogarasi (1997); c: Kobayashi (1998); d: Colominas et al. (1996) | |
| Table 3: | Dipole moments (Debye) of the amino-oxo (compound 1) and amino-hydroxy (compound 2) obtained in this work and in other theoretical and experimental results |
a: Present study; b: Clowney et al. (1999); c: Kulakowska et al. (1975); d: Balkalarski et al. (1996) | |
Similarities in structures are indicated by similarities in the dipole moments. The dipole moment obtained in this work for amino-oxo (1) is in the range of the experimental (Kulakowska et al., 1975; Balkalarski et al., 1996) values and not too far from some values obtained by MP2 and B3LYP (Clowney et al., 1999) methods. No experimental values are found for the amino-hydroxy tautomer (2), (Table 3).
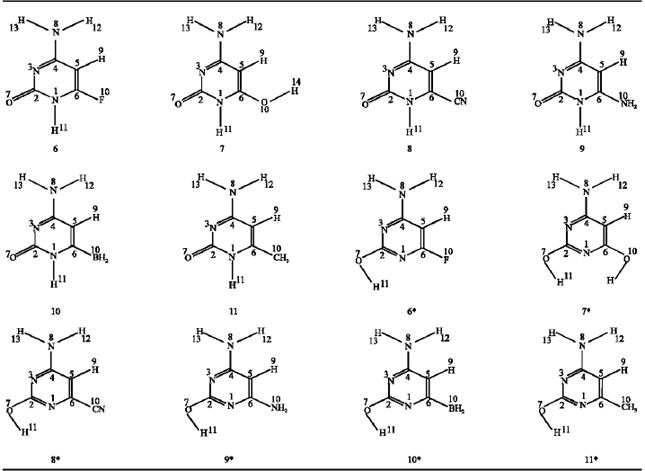
| Table 4: | Substituted amino-oxo and amino-hydroxy compounds |
 | |
| Table 5: | Selected bond lengths and bond angles (Å and deg.) for the substituted amino-oxo cytosines |
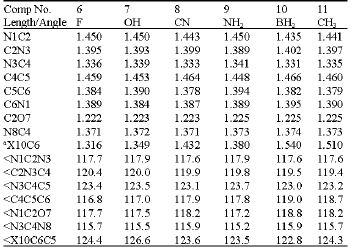 | |
a: For atom X see the corresponding structure from (Table 4) | |
Effect of substituents: The F, OH, CN, NH2, BH2 and CH3 substituents were introduced at position 6 to both amino-oxo (1) and amino-hydroxy (2) compounds, these structures are shown in (Table 4).
The geometrical parameters (bond lengths and bond angles) for these substituted amino-ox and amino-hydroxy cytosines are shown in Table 5 and 6, respectively. .
| Table6: | Selected bond lengths and bond angles (Å and deg.) for the substituted amino-hydroxy cytosines |
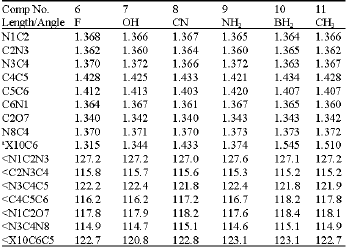 | |
a: For atom X see the corresponding structure from (Table 4) | |
The variation in bond lengths for all bonds before and after substitution in the case of amino-oxo and amino-hydroxy cytosines was calculated. It is found that the C2O7 and N8C4 bond lengths were almost constant where the change was between 0.000 and 0.002 Å. The effect of the CH3-group substitution on the amino-oxo and F-group substitution on the amino-hydroxy cytosines was the minimum compared to the other substituents. Also, it can be noticed, generally, that the bond lengths at the substitution position (C5C6 and C6N1) were the most affected ones compared to the other bond lengths in the substituted amino-oxo and amino-hydroxyl cytosines
The effect of substituents on the equilibrium position has been studied by calculation of the free energy change and the enthalpy change for the appropriate isodesmic reactions.
Free energy change
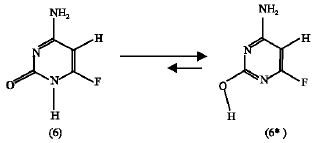
Effect of F substituent: The calculated heat of formation of the amino-oxo F-substituted sytosine (6) is -248.59 kJ mol1 and that of the amino-hydroxy F-substituted cytosine (6*) is -294.88 kJ mol1. Also, the entropy of the former compound is 58.43 J mol1 K and that of the later is 57.08 J mol1 K. So, at 25C the free energy change ΔG for the reaction in scheme 4 below is -44.62 kJ mol1.
ΔG = -44.62 kJ mol1 | (2) |
 |
| scheme 4 |
The more negative ΔG value compared to that of the unsubstituted system in scheme 3 means that the F substitution increases the tendency of the reaction to happen in the forward direction.
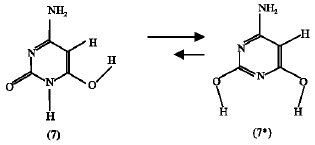
Effect of OH substituent: The heat of formation of the OH-substituted amino-oxo cytosine (7) is found to be -262.61 kJ mol1 and that of OH-substituted amino-hydroxy cytosine (7*) is found to be -317.99 kJ mol1. This means that the OH-substituted amino-hydroxy (7*) is more stable than the OH-substituted amino-oxo (7) by 55.38 kJ mol1. The absolute entropy values are found to be 248.15 and 241.77 J mol1 K for compounds (7) and (7*), respectively. Accordingly, the calculated free energy change ΔG for the tautomerization reaction (scheme 5) at 25C is -53.47 kJ mol1 So, the substitution of OH group at position 6 increases the tendency of the reaction to happen in the forward direction compared to the unsubstituted system (scheme 3) where ΔG becomes more negative.
ΔG = -53.47 kJ mol1 | (3) |
 |
| scheme 5 |
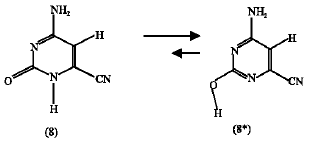
Effect of CN substituent: The heats of formation of the CN-substituted amino-oxo (8) and amino-hydroxy (8*) cytosines were found to be 95.43 and 46.28 kJ mol1 respectively. The amino-hydroxy form has lower heat of formation so it is more stable compared to the amini-oxo form by 49.15 kJ mol1. The entropy values were found to be 259.17 J mol1 K for the amino-oxo (8) form and 255.68 J mol1 K for the amino-hydroxy (8*) form. The calculated free energy change (ΔG) at 25C according to these data is -48.11 kJ mol1. This means that the reaction (scheme 6) favors the forward direction, also it can be said that the CN-substituent at position 6 increases the tendency of the reaction to happen in the forward direction because of the lower free energy change value obtained after substitution. The ΔG value before substitution was -39.44 kJ mol1.
ΔG = -48.11 kJ mol1 | (4) |
 |
| scheme 6 |
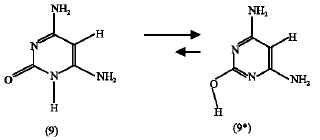
Effect of NH2 substituent: The calculated heat of formation of the NH2-substituted amino-oxo (9) and amino-hydroxy (9*) cytosines were -42.62 and -96.52 kJ mol1, respectively. The entropies obtained were 248.92 J mol1 K for compound (9) and 242.52 J mol1 K for compound (9*). From these data the calculated ΔG for the tautomerization reaction (schem 7) is -51.99 kJ mol1. This value is negative and lower than that obtained from the unsubstituted reaction in scheme 3. So, NH2 group substitution also encourages the forward reaction and increases the tendency of the reaction in the forward direction compared to the unsubstituted tautomerization reaction (scheme 3).
ΔG = -51.99 kJ mol1 | (5) |
 |
| scheme7 |
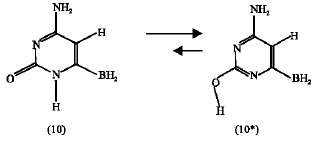
Effect of BH2 substituent: The heat of formation obtained for the BH2-substituted amino-oxo cytosine (10) is -22.99 kJ mol1 and that of it`s counter amino-hydroxy cytosine (10*) is -61.03 kJ mol1. This means that compound (10*) is 38.04 kJ mol1 more stable than compound (10). The absolute entropy for compound (10) is 255.99 J mol1 K and that of (10*) is 253.44 J mol1 K. The calculated free energy change (ΔG) for the tautomerization reaction (scheme 8 ) at 25°C therefore is -37.28 kJ mol1. This value of free energy change is 2.16 kJ mol1 higher than that of the unsubstituted tautomeric pair (scheme 3). This means that the BH2-substituent has decreased the tendency of the tautomerization reaction in scheme 8 to happen in the forward reaction compared to the unsubstituted pair (scheme 3) but still the reaction favors the forward direction because it is highly energetically favorable where ΔH is -38.04 kJ mol1.
ΔG = -37.28 kJ mol1 | (6) |
 |
| scheme 8 |
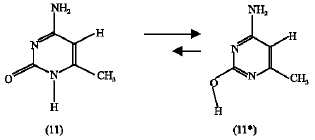
Effect of CH3 substituent: The calculated heat of formation of the CH3-substituted amino-oxo cytosine (11) and that of CH3-substituted amino-hydroxy cytosine (11*) are found to be -82.17 and -126.53 kJ mol1, respectively. This means that the later is more stable than the former by 44.36 kJ mol1 and therefore the product (11*) is energetically favorable over the reactant (11), scheme 9 . The entropy change was found to be -4.70 J mol1 Kand therefore the free energy change (ΔG) is -42.96 kJ mol1 at 25C and the forward direction is still favorable.
ΔG = -42.96 kJ mol1 | (7) |
 |
| scheme 9 |
For all of the substituents it is noticed that the entropy changes for the tautomerization reactions were negative which favors the backward reaction. But these values are small such that at 25C the entropy-factor (TΔS) effect on the free energy change (ΔG) was small compared to the effect of the energy factor (ΔH) which is largely negative, so, the reactions are pushed in the forward direction under the effect of energy factor.
It is important not to forget that thermodynamics is not sufficient to have a full idea about reactions, i.e, the kinetics must be taken into consideration. ΔG values may be largely negative but the kinetics may imply very slow reaction, i.e., long time to reach equilibrium. A work done by Podolyan and co-workers emphasized the importance of the kinetics in ab initio study on the contribution of cytosine and guanine tautomerism on the spontaneous mutation (Podolyan et al., 2003).
Isodesmic reactions: The effect of the substituents on both amino-oxo and amino-hydroxy forms of cytosine is also studied by isodesmic reactions. The results obtained from isodesmic reactions support those obtained from the free energy calculations. They will be exemplified by F, NH2 and CH3 substituents because they represent different cases regarding the combinations of enthalpy changes for the isodesmic reactions of the substituted amino-oxo and amino-hydroxy cytosines. Positive enthalpy change for the isdesmic reaction means that the substituent stabilizes the compound, where as a negative enthalpy change indicates a destabilization effect (Lindner and Lemal, 1996). Also, more positive values indicate more stabilization and more negative values indicate more destabilization as illustrated in the introduction part.
Effect of F substituent: The enthalpy change for the isodesmic reaction of the F-substituted amino-oxo cytosine (6) is -1.979 kJ mol1 (reaction 8) and that of F-substituted amino-hydroxy cytosine (6*) is 0.113 kJ mol1 (reaction 9). The negative value of the enthalpy change for the F-substituted amino-oxo form of cytosine (6) mean that the F substitution has destabilization effect on this compound (6) but it has a stabilization effect of the amino-hydroxy cytosine (6*) because the enthalpy change for the corresponding isodesmic reaction (reaction 9) is positive. Therefore, it can be said that F-substitution encourages the product (6*) formation or shifts the reaction (scheme 4) in the forward direction. This result supports that obtained by the free energy calculations where the ΔG value obtained was negative.
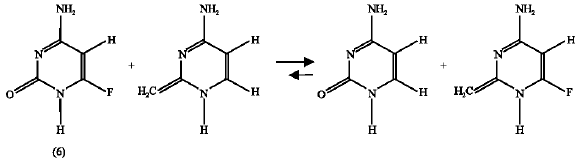 | (8) |
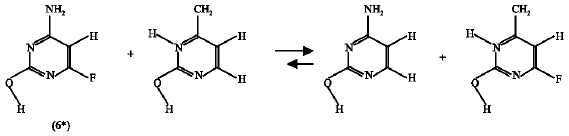 | (9) |
Effect of NH2 substituent: For this substituent, it is found that the enthalpy change for the isodesmic reaction of HN2-substituted oxo-amino cytosine (9) (reaction 10) is 5.552 kJ mol1 and that of the substituted amino-hydroxy parent (9*) (reaction 11) is 10.486 kJ mol1. This means that the NH2 substituent stabilized both the amino-oxo (9) and the amino-hydroxy (9*) cytosines but the later is stabilized more because the enthalpy change of it`s corresponding isodesmic reaction is higher.
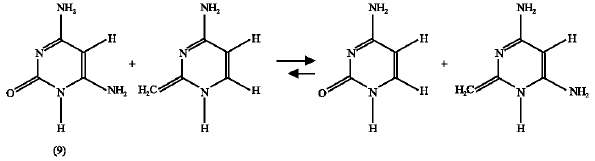 | (10) |
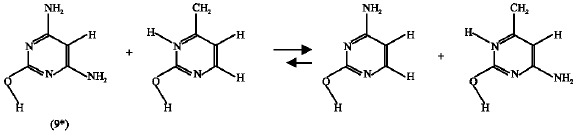 | (11) |
So, from the isodesmic reactions` enthalpy-change calculations it is concluded that the product (9*) is more stable than the reactant (9) and the forward reaction (scheme7 ) is favorable. This result supports that obtained earlier by the free energy change calculations.
Effect of CH3 substituent: In this case, the enthalpy change for the isodesmic reactions corresponding the CH3-substituted amino-oxo (11) (reaction 12) and amino-hydroxy (11*) (reaction 13) were -0.198 and -0.106, respectively.
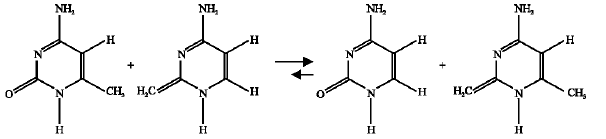 | (12) |
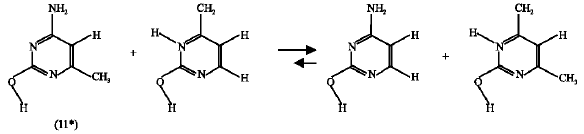 | (13) |
These negative values mean that the CH3 substituent destabilized both the amino-oxo (11) and it`s amino-hydroxy (11*) counterpart, but the amino-oxo (11) is more destabilized because it`s corresponding isodesmic reaction has lower enthalpy change value. This suggests that the reactant (11) is more destabilized than the product (11*), i.e., the forward direction of the reaction (scheme 9 ) is favorable. The same result was obtained from the free energy calculations.
CONCLUSION
Calculations performed on 14 tautomers and rotamers of cytosine in it`s different forms of amino-oxo, amino-hydroxy, imino-oxo and imino-hydroxy showed that the amino-hydroxy (2) is the most stable tautomer. None of the substituents studied in this work was able to reverse the equilibrium towards the amino-oxo direction as indicated by the Gibbs free energy and isodesmic reacations` calculations.
REFERENCES
- Balkalarski, G., P. Grochowski, J.S. Kwiakowski, B. Lesyng and J. Leszczynski, 1996. Molecular and electrostatic properties of the N-methylated nucleic acid bases by density functional theory. Chem. Phys., 204: 301-311.
CrossRef - Brown, R.D., P.D. Godfry and D. McNaughton, 1989. Tautomers of cytosine by microwave spectroscopy. J. Am. Chem. Soc., 111: 2308-2810.
CrossRefDirect Link - Chung, G., H.B. Oh and D. Lee, 2005. Tautomerism and isomerism of guanine-sytosine DNA base pair: Ab initio and density functional theory approaches. J. Mol. Struct. Theochem., 730: 241-249.
CrossRef - Civcir, P.U., 2000. Atheoretical study of tautomerism of cytosine, thymine, uracil and their 1-methyl analogues in the gas and aqueous phases using AM1 and PM3. J. Mol. Struct. Theochem., 532: 157-169.
CrossRef - Clowney, L., J.D. Westbrook and H.M. Berman, 1999. CIF applications. XI. A La Mode: A ligand and monomer object data environment. 1. Automated construction of mm CIF monomer and ligand models. J. Applied Crystalogr., 32: 125-133.
CrossRefDirect Link - Colominas, C., F.J. Luque and M. Orozco, 1996. Tautomerism and protonation of guanine and cytosine, implications in the formation of hydrogen-bonded complexex. J. Am. Chem. Soc., 118: 6811-6821.
CrossRefDirect Link - Cysewski, P., 2005. An ab initio study on nucleic acid bases aromaticities. J. Mol. Struct. Theochem., 714: 29-34.
CrossRef - Dewar, M.J. and W. Thiel, 1977. Ground states of molecules. 38. The MNDO method of approximations parameters. J. Am. Chem. Soc., 99: 4899-4907.
CrossRefDirect Link - Estrin, D.A., L. Paglieri and G. Corongiu, 1994. A density functional study of tautomerism of uracil and cytosine. J. Phys. Chem., 98: 5653-5660.
CrossRefDirect Link - Fazaeli, R., M. Moajjemi, F. Ataherian and K. Zare, 2002. Solvent effects on relative and 14N NMR shielding of cytosine tautomrs: Contiuous set of gauge transformation calculation using polarizable continuum model. J. Mol. Struct. Theochem., 581: 51-58.
CrossRef - Fogarasi, G., 1997. High-level electron correlation calculations on some tautomers of cytosine. J. Mol. Struct., 413-414: 271-278.
CrossRef - Fogarasi, G. and P.G. Szalay, 2002. The interaction between cytosine tautomers and water: An MP2 and coupled cluster electron correlation study. Chem. Phys. Lett., 356: 383-390.
CrossRef - Gould, I.R., M.A. Vincent, I.H. Hiller, L. Lapinski and M.J. Nowak, 1992. A new theoretical prediction of the infrared spectra of cytosine tautomers. Spectrochim. Acta Part A: Mol. Spectroscopy, 48: 811-818.
CrossRef - Hehre, W.J., R. Ditchfield, L. Radom and J. A. Pole, 1970. Molecular orbital theory of the electronic structure of organic compounds.V. molecular theory of band separation. J. Am. Chem. Soc., 92: 4796-4801.
Direct Link - Jaworski, A., M. Szczepaniak, K. KiBulat and W.B. Person, 1990. Infrared spectra and tautomerism of 5-fluorocytosine, 5-bromocytosine and 5-iodocytosine. Matrix isolation and theoretical AB initio studies. J. Mol. Struct., 223: 63-92.
CrossRef - Kobayashi, R., 1998. A CCSD(T) study of the relative stabilities of cytosine tautomers. J. Phys. Chem., 102: 10813-10817.
CrossRefDirect Link - Kulakowska, M.G., B. Lesyng, K.L. Wierzchowski and K. Bolewska, 1975. Barrier to rotation and conformation of the -NR2 group in cytosine and its derivatives. Part II. Experimental and theoretical dipole moments of methylated cytosines. Biochim. Biophys. Acta, 407: 420-429.
PubMedDirect Link - Les, A. and L. Adamowicz, 1989. Oxo-hydroxy tautomerism of uracil and 5-fluorouracil. J. Phys. Chem., 93: 7078-7081.
CrossRefDirect Link - Lindner, P.E. and D.M. Lemal, 1996. Highly fluorinated cyclopentanones and their enoles. J. Org. Chem., 61: 5109-5115.
CrossRefDirect Link - Nowak, M.J., L. Lapinski and J. Fulara, 1989. Matrix isolation studies of cytosine: The separation of the infrared spectra of cytosine tautomers. Spectrochim. Acta Part A: Mol. Spectroscopy, 45: 229-242.
CrossRef - Orozco, M., B. Hernandez and F.J. Luque, 1998. Tautomerism of 1-methyl derivatives of uracil, thymine and 5-bromuracil, is tautomerism the basis for mutagenicity of 5-bromouridine? J. Phys. Chem. B, 102: 5228-5233.
CrossRefDirect Link - Piacenza, M. and S. Grimme, 2003. Systematic quantum chemical study of DNA-base tautomers. J. Comp. Chem., 25: 83-99.
CrossRefDirect Link - Podolyan, Y., L. Gorb and J. Leszczynski, 2003. Ab initio study of the prototropic tautomerism of cytosine and guanine and their contribution to spontaneous point mutation. Int. J. Mol. Sci., 4: 410-421.
CrossRefDirect Link - Rueda, M., F.J. Luque, J.M. Lopez and M. Orozco, 2001. Amino-imino tautomerism in derivatives of cytosine: effect on hydrogen-bonding and stacking properties. J. Phys. Chem. A, 105: 6575-6580.
CrossRefDirect Link - Sambrano, J.R., A.R. de Souza, J.J. Queralt, M. Oliva and J. Andres, 2001. Density functional study of the 5-methylcytosine tautomers. Chem. Phys., 264: 333-340.
CrossRef - Sambrano, J.R., A. R. de Souza, J.J. Queralt and J. Andres, 2000. A theoretical study on cytosine tautomers in aqueous media by using continuum models. Chem. Phys. Lett., 317: 437-443.
CrossRef - Scanlan, M. and I.H. Hillier, 1984. Ab initio study of tautomerism of uracil, thymine, 5-fluorouracil and cytosine. J. Am. Chem. Soc., 106: 3737-3745.
CrossRefDirect Link - Topal, M.D. and J.R. Fresco, 1976. Base pairing and fidelity in codon-anticodon interaction. Nature, 263: 289-293.
CrossRefDirect Link - Voet, D. and A. Rich, 1970. The crystal structures of purines, pyrimidines and their intermolecular complexes. Prog. Nucleic Acid Res. Mol. Biol., 10: 183-265.
CrossRef - Wolken, J.K., C. Yao, F. Turecek, M.J. Polce and C. Wesdemiotis, 2007. Cytosine neutral molecules and cation-radicals in the gas-phase structures, energetics, ion chemistry, and neutralization-reionization mass spectroscopy. Int. J. Mass Spectrometry, 267: 30-42.
CrossRef - Yao, C., F. Turecek, M.J. Polce and C. Wesdemiotis, 2007. Proton and hydrogen atom adducts to cytosine, an experimental and computational study. Int. J. Mass Spectrometry, 265: 106-123.
CrossRef - Zhao, Z., Q. Zhang, C. Gao and Y. Zhuo, 2006. Motion of the hydrogen bond proton in cytosine and the transition between it`s normal and imino states. Phys. Lett., A359: 10-13.
CrossRef