
C.S. Sipaut
School of Chemical Sciences, Universiti Sains Malaysia, 1 1 800 P. Pinang, Malaysia
N. Ahmad
School of Chemical Sciences, Universiti Sains Malaysia, 1 1 800 P. Pinang, Malaysia
R. Adnan
School of Chemical Sciences, Universiti Sains Malaysia, 1 1 800 P. Pinang, Malaysia
I.Ab. Rahman
School of Chemical Sciences, Universiti Sains Malaysia, 1 1 800 P. Pinang, Malaysia
M.A. Bakar
School of Chemical Sciences, Universiti Sains Malaysia, 1 1 800 P. Pinang, Malaysia
J. Ismail
School of Chemical Sciences, Universiti Sains Malaysia, 1 1 800 P. Pinang, Malaysia
C.K. Chee
1nte1 Technology (M) Sdn. Bhd, Bayan Lepas FIZ Phase 111, 1 1900 P. Pinang, Malaysia
Journal of Applied Sciences
Year: 2007 | Volume: 7 | Issue: 1 | Page No.: 27-34







ABSTRACT
Modified fumed silica-epoxy nanocomposites were obtained by refluxing epoxy molecule with fumed silica using imidazole as catalyst. The modified fumed silica was then used as filler in epoxy resin with amine as curing agent. The properties of the surface modified silica and their effect as fillers in bulk epoxy composite were characterized by Fourier Transform Infrared spectroscopy (FTIR), Proton Nuclear Magnetic Resonance Spectroscopy (1H-NMR), Thermogravimetri Analysis (TGA), Differential Scanning Calorimeter (DSC), Coefficient of Thermal Expansion (CTE), Tensile testing, Scanning Electron Microscopy (SEM) and Energy Dispersive X-ray spectrometry (EDX). From FTIR, 1H-NMR and TGA analysis, it was found that the epoxy resin was chemically bonded onto silica surface. From the DSC and CTE analysis, the addition of modified silica filler in the composite matrix highly influences thermal properties. This new synthesis filler shows higher glass transition temperature and more stable CTE data compared to unmodified filler when introduce into composite matrix. The tensile properties of composite matrix with and without the addition of filler show no significant difference in their tensile properties. SEM-EDX analysis show modified fillers have better adhesion with composite matrix compared to unmodified filler.
PDF Abstract XML References Citation
How to cite this article
C.S. Sipaut, N. Ahmad, R. Adnan, I.Ab. Rahman, M.A. Bakar, J. Ismail and C.K. Chee, 2007. Properties and Morphology of Bulk Epoxy Composites Filled with Modified Fumed Silica-Epoxy Nanocomposites. Journal of Applied Sciences, 7: 27-34.
DOI: 10.3923/jas.2007.27.34
URL: https://scialert.net/abstract/?doi=jas.2007.27.34
DOI: 10.3923/jas.2007.27.34
URL: https://scialert.net/abstract/?doi=jas.2007.27.34
INTRODUCTION
Epoxy composites with silica are normally used in encapsulation materials for semiconductor devices (Zheng and Ning, 2003; Chen et al., 2004; Ritzenthaler et al., 2000; Ho and Wang, 2001; Wang et al., 2002). The used of silica in semiconductor provide significant properties changes in the epoxy composite. Normally, silica provides high thermal stability, strength, hardness and low cost. These combinations strongly can achieve the high performance in electrical applications (Bauer et al., 2004; Kickelbick, 2003; Becker et al., 2003; Salahuddin et al., 2002).
Fumed nanosilica based materials are normally low cost and it can be manufactured by high-temperature hydrolysis of silicon tetrachloride in a flame producing smaller sizes 15-30 nm (Wu et al., 2005). The silanol and siloxane group are covered on the silica surface, leading to many applications of silica such as rheological controlling agent in epoxy resin systems, as fillers in toothpaste and car tires and as starting material for optical fibers (Barthel, 1995; Shirono et al., 2001; Stesmans et al., 2005).
However, the intrinsic properties, namely interaction between organic and inorganic phase and dispersibility of the filler in organic media largely affect the properties of composite matrix. Therefore, for excellent properties, strong interfaces between components are required that are normally achieved by the introduction of silica with modification using some organic molecules (Kang et al., 2001).
Kang et al. (2001) reported that in the preparation of epoxy composites filled with functionalized nanosilica particles, surface modified particles highly affect the particle dispersion and interface in bulk epoxy composite. They investigated the behavior the Coefficients of Thermal Expansion (CTE) and glass transition temperature (Tg) of bulk epoxy composite with different silica contents and found that the CTE of bulk epoxy composites are reduced and Tg increased with increasing filler contents.
The compatibility effect of poly(propylene-g-maleic anhydride) copolymer (PP-g-MA) on isotactic poly(propylene)-silica nanocomposites on the morphology behavior and mechanical properties of the nanocomposites has been studied by Bikiaris et al. (2005). The authors indicate that the addition of modified silica particles result in enhance mechanical properties, compared with that of unmodified silica particles, due to the higher silica dispersion in the polymer matrix.
However, the study conducted using modified fumed nanosilica with surface modification and at smaller sizes (15-30 nm) has limited study. Most of the investigation generally focused on colloidal silica with size in the range of 10 to 20 nm (Liu et al., 2003a,b).
Sipaut et al. (2005) reported that fumed nanosilica particles were successfully modified by epoxy molecule. This study reports our findings on the physical and chemical effects and mechanical properties of the modified and unmodified nanosilica filler in the composite system.
MATERIALS AND METHODS
Materials: Nanoscale silica particles used was fumed nanosilica (99.8% purity), manufactured by Sigma with the mean particle sizes in the range of 15-30 nm. Diglycidyl ether of bisphenol A (modifying agent) was used as the surface modifier and was manufactured by Durcupan ACM. Reaction catalyst used was imidazole manufactured by Fluka.
Epoxy-composite resin used was prepared using epoxy resin clear 331 (bisphenol type) manufactured by Echemo Trading with an average molecular weight of 700 g mol–1. The amine hardener (Epoxy hardener clear 2963) of Echemo Trading, is a mixture of trimethylhexamethylene diamine (5-11%), isophorone diamine (30-42%) and benzyl alcohol (30-42%). This study was conducted in School of Chemical Sciences, Universiti Sains Malaysia from July 2004 until May 2005.
Sample preparation:
Surface modification of fume silica: Mixtures of 40% of fumed silica in modifying agent were dispersed in 50 mL toluene in the presence of imidazole (25%). The mixture was then refluxed at 100°C for 2 h. To remove byproduct, centrifuging technique were conducted at least three times using acetone as solvent. The product was then further dispersed in 50 mL acetone and stirred at room temperature for another 3 h. The collected product was then further centrifuged and dried at 110°C in an oven. The Modified Silica (MS) nanoparticles were pretreated at 110°C under vacuum for 24 h to eliminate water on the particles surface.
Epoxy composite preparation: In matrix composite preparation, epoxy resin 331 and amine hardener 2963 was mixed at 1:1 ratio. The mixture was then stirred for 10 min and poured into preheated polytetrafluoroethylene (PTFE) mould and cured at room temperature for 5 h. The addition of modified or unmodified fumed nanosilica filler in the composite matrix was prepared by dispersing the filler into amine hardener 2963 at 50±2°C using ultra sonic instrument for 2 h. The sample was then gently mixed with epoxy resin 331 to minimize air bubbles formation and cured at room temperature for 5 h.
Characterization: All samples either the modified or unmodified nanosilica particles were subjected to chemical analysis to identify the chemical interaction in the modification stage. The chemical analysis includes Fourier transform infrared (FTIR) measurements were performed on Pelkin Elmer 2000 in the 4000 to 400 cm–1 region with 4 cm–1 resolution. A small amount (~0.004 g) of sample was thoroughly mixed with 0.4 g at dried KBr and pressed to obtain a cohesive transparent disc. The sample disc was then place in a specific sample holder in the IR-instrument with background scan of KBr before measurements. Proton nuclear magnetic resonance (1H-NMR) was also conducted on an Advance 400 MHZ Bruker FT-NMR Spectrometer in DMSO-d6. Thermogravimetri analysis (TGA) was performed using a Mettler Toledo (TGA/SDTA 851e) under nitrogen flow at heating rate of 10.0°C min–1 from 50 to 950°C.
All samples of bulk epoxy composite either with or without the addition of modified or unmodified nanosilica filler were subjected to selected analysis. Differential Scanning Calorimetry, DSC was performed using Pelkin Elmer Pyris 1 DSC. Samples of 5 mg were sealed in aluminum pans from-50 to 300°C at a scanning rate of 20°C min–1 under nitrogen atmosphere. At least five separate samples were tested and results were quoted as a mean value. The Coefficient of Thermal Expansion measurement (CTE) was measured with a dilatometer (NETZSCH DIL 402 C) in the temperature range of 30 to 60°C. A sample of approximately 2 mm in length was used at heating rate of 10°C min–1 under nitrogen atmosphere. The mechanical properties (tensile) was determined using a Hounsfield Test Equipment model using a cross head speed of 100 mm min–1 with a gauge length was set at 40 mm. Samples with a dimension of 80x10x2 mm3 were used in this analysis. The result was quoted as the mean of five separate samples of each formulation.
The morphology of modified and unmodified filler in the composites matrix was examined by Leo Supra 50 VP Field Emission model SEM. All samples were gripped by pliers, chilled in liquid N2 and then fractured. The specimen was deposited on double-sided scotch and was examined on the fractured surface. The fractured surfaces were then coated with thin layer (20 nm) of gold to improve Scanning electron microscope, SEM imaging using a Polaron SC 515. Energy Dispersive X-ray spectrometry, EDX analysis was also used to determine the chemical elements compositions in the samples using an Oxford INCA 400 energy dispersive x-ray microanalysis system attached to SEM instrument.
RESULTS AND DISCUSSION
Modified fumed silica:
Analysis of modified silica by FTIR: The analysis was conducted to compare the changes in functional groups on the fumed nanosilica surface before and after modification. Figure 1 showed the FTIR spectra of pure nanosilica (PS) and modified nanosilica (MS).
From the spectra, it shows that there is a significant new peak appeared for the MS compared to PS. The new peaks, such as weak peaks near 2850 to 2960 cm–1 and 3000 to 3100 cm–1 correspond to the absorption of C-H stretching (aliphatic and aromatic ring) from epoxy molecules. Moreover, the absorption peak of hydroxyl groups at 3405 cm–1 is broader indicating that the presence of new hydroxyl groups coming from the ring opening reaction of the epoxide ring. Peaks appearing at 1509 to 1582 cm–1 and 745 to 771 cm–1 correspond to aromatic C=C stretching and ortho-disubstituted benzene ring respectively. The weak band at 1384 and 1362 cm–1 are due to-CH3 stretching of epoxy molecule. C-O stretching of the epoxy appears at 1297 and 1181 cm–1. Strong C-H bending, out of plane band occurs at 830 to 971 cm–1. All of this is attributed to the epoxy modifier molecules.
Based on the appearance of these new peaks, it was strongly suggested that epoxy modifier is chemically bonded with fumed silica. This phenomenon was further investigated by 1H-NMR analysis.
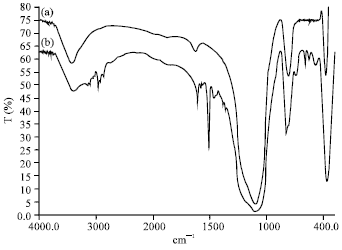 | |
| Fig. 1: | FTIR spectra of (a) PS and (b) MS |
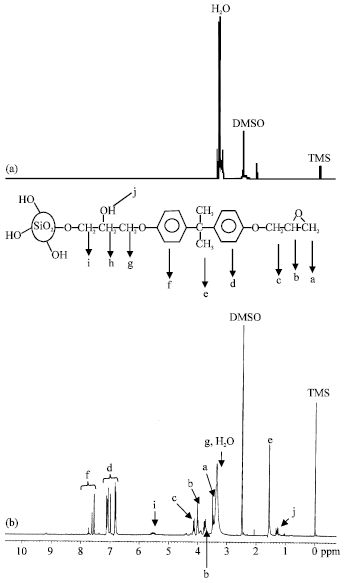 | |
| Fig. 2: | 1H-NMR spectra of (a) PS and (b) MS |
 | |
| Fig. 3: | TGA spectra of (a) PS and (b) MS |
2. 1H-NMR interpretation of the modified silica: This analysis was carried out on samples with unmodification and modification process. Figure 2 shows the 1H-NMR spectra for PS and MS samples.
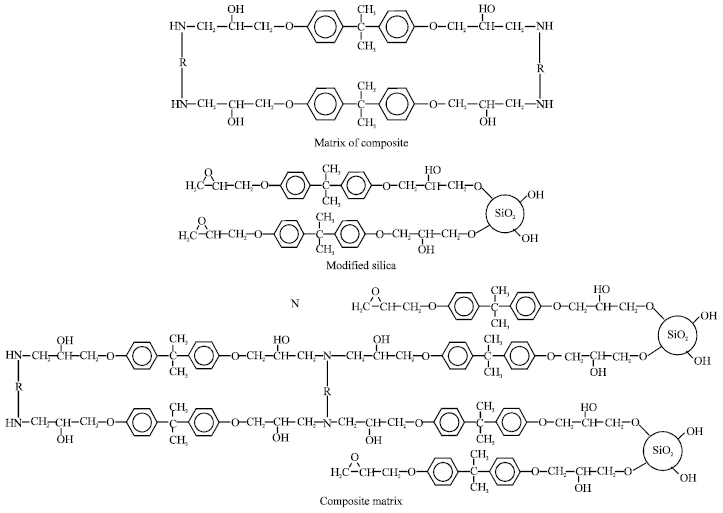 | |
| Fig. 4: | Formation of composite matrix |
From the spectra, two peaks at 2.5 and 3.4 ppm in PS sample are attributed to H2O and DMSO solvent respectively (Fig. 2a). However, Fig. 2b (i.e., MS sample) shows additional peaks compared to PS sample. From Fig. 2b, a doublet at ~3.4 ppm (peak a) was attributed to-CH-(O)-CH2 group, a multiplet at 3.4 to 3.6 ppm (peak b) is assumed to be-CH-(O)-CH2) group and the doublet at ~ 4.1 ppm (peak c) corresponds to-OCH2-CH-group indicating the presence of an oxirane ring. The signals at 6.8 to 7.1 ppm (peak d) and 7.5 to 7.6 ppm (peak f) region correspond to the presence of two different aromatic protons.
The formation of silanoxymethylene (Si-O-CH2) group was observed with the appearance of a doublet at ~ 1.3 ppm (peak j). This peak signifies the reaction of Si-OH and the oxirane ring from the epoxy. Moreover, a singlet at ~ 5.5 ppm (peak I) is attributed to-CH2CH(OH)-CH2-group while the multiplet at 3.7 to 4.0 ppm (peak h), indicates the presence of the-CH2CH(OH)-CH2-group from ring opening reaction of the oxirane.
A singlet at ~1.6 ppm (peak e) is attributed to-CH4-C(CH3)2-CH4-group and the doublet at ~3.4 ppm (peak g) is assumed to correspond to the-CH-CH2-O-group. This 1H-NMR observation again supports the findings that epoxy molecules were chemically bonded onto the fumed silica surface.
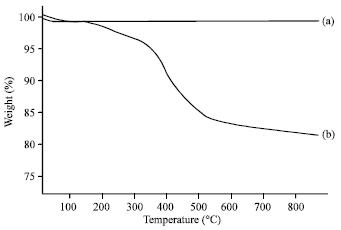
TGA analysis on modified silica: TGA analysis on modified nanosilica, Fig. 3, shows the TGA thermogram of PS and MS. TGA thermogram of PS shows no weight loss during the heating temperature from 50 to 950°C and also no release of any other substances which is in accordance with Liu et al. (2004). However, MS is stable up to only 200°C. A drop is noted at above 200°C up to 650°C with a total weight loss of 18.22%. The weight loss was expected to be due to the organic groups because the thermal decomposition of the epoxy resin normally occurs between 200 to 400°C. Again this observation supports the findings that the nanosilica surface was successfully modified with epoxy modifier (Liu et al., 2004).
To observe the behavior of the modified nanosilica with epoxy molecule as filler in the composite matrix, 5% of the filler was introduced into the composite matrix and were compared with the unmodified silica sample.
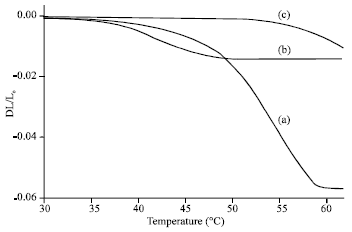 | |
| Fig. 5: | dL/Lo of (a) Blank composite, (b) Composite with 5% PS content and (c) Composite with 5% MS content on increasing temperature |
| Table 1: | Tg value of blank, 5% PS and 5% MS composite matrix |
 | |
| Table 2: | Tensile properties of blank, 5% PS and 5% MS composite matrix |
 | |
The composite matrix was then analyzed by DSC, CTE, mechanical test and SEM-EDX.
The effect of modified silica as filler in composite matrix
The effect of glass-transition temperature: DSC was completed on the unfilled and filled samples with 5% fillers. Tgs of the samples were determined from the tangents of DSC spectra as a function of temperature. Table 1 provides information related to the Tg value of epoxy composites with and without the addition of 5% PS and MS. The Tg value of composite matrix in the absence of filler is slightly lower than composite in the presence of 5% PS and MS filler. The lower Tg value of composite matrix in the absence of filler compared to with PS and MS filler is attributed to the formation of chemical and physical crosslink either by hydrogen bonding formation between silica surface or covalent bonding in the matrix composite.
Furthermore, comparing the Tg value between MS and PS, it shows that composite prepared with 5% of MS filler has higher Tg than with 5% of PS filler. The higher value of Tg for MS filler composite possibly due to increase in the formation of covalent bond between silica surface and polymer matrix to form crosslink compared to the other system. This behavior can be explained using free volume concept. The increase of cross linking in polymer matrix will reduce the specific volume and less molecular motion required more energy for rotation therefore increase Tg value. This finding not only shows the improvement of the Tg values in the composite but also supported the successfulness of the modification of the silica surface with epoxy molecule modifier (Young and Lovell, 1991). The possibility of covalent bonding formation between the MS with epoxy composite can be illustrated by chemical reaction (Fig. 4).
The effect of CTE: In this study, the CTE value was measured as the changes in sample length (as the temperature increased) divided with the original sample length at room temperature. Figure 5 shows the CTE value, (dL/Lo), of blank, PS and MS composite matrix with increasing temperature up to 60°C. The result shows that for blank composite sample, the dL/Lo value start to drop to negative value at approximately 40°C. The dL/Lo value tends to show higher negative value as temperature is increased. The decreased in dL/Lo i.e., CTE values (negative value) suggested that the sample have a tendency to shrink i.e., unstable sample. Sample prepared with PS filler also show drops in dL/Lo value at 40°C. However the margin of dL/Lo value is less that the blank composite. This hinted that the addition of PS filler in the composite matrix reduce the degree of shrinkage.
Furthermore the MS composite sample exhibit dropped in dL/Lo value at 52°C and the degree of shrinkage is lower than PS and blank composite sample. This highly suggested that the modified nanosilica filler increase the temperature stability and provided lower degree of shrinkage compared to unmodified filler. This is possibly due to the covalent bonding formation between the filler with the composite matrix providing better interaction resulting difficulties to shrink or expand.
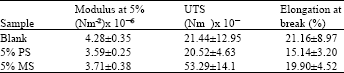
Mechanical properties: Table 2 shows that modulus at 5%, ultimate tensile strength, UTS and elongation at break value with blank, PS and MS bulk epoxy composite. From the results it shows that within the limit of experimental error there is no significant different on the tensile values of all formulations. This hinted that the samples prepared possibly have uneven surface or not homogeneously mixed. However, in the presence of either PS or MS filler, the distributions of filler tend to form agglomerate (i.e., not a good dispersion) in the composite matrix. This was supported by Xing and Li (2004) who reported that lower percentage of filler (below 2.5%) in the composite matrix give good dispersion however higher filler loading (above 4%) tends to form agglomerate.
 | |
| Fig. 6: | SEM micrographs on fracture surface of (a) unfilled composite matrix, (b) unmodified filler composite matrix and (c) modified filler composite matrix |
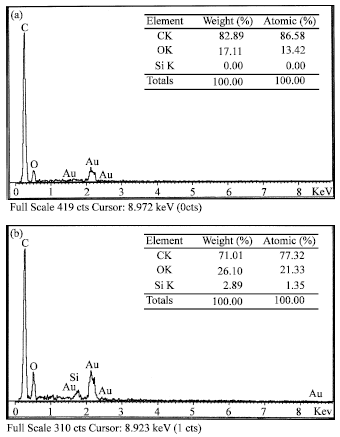 | |
| Fig. 7: | EDX spectra of (a) 5% PS composite and (b) 5% MS composite |
Nevertheless, this data suggest that to improve the mechanical analysis, the preparation of the sample must be improved. Furthermore, for composite sample with presence of a filler, the loading capacity need to be less than 1% to determine the changes in the tensile properties (Wu et al., 2002).
SEM-EDX analysis of fractured surface: SEM analysis was conducted to investigate the dispersion and interfacial properties in bulk epoxy composite matrix on fractured surfaces. SEM photograph of blank epoxy composite (in the absence of filler) shows the clear river lines with a smooth surface on the fracture surface part (Fig. 6a). However Fig. 6b shows the SEM micrograph of composite 5% PS which illustrates more river lines than Figure 6(a). This suggested that the additions of PS into epoxy matrix highly affect the fracture surface and brittle behavior of the composite matrix.
This phenomenon is attributed to the formation of hydrogen bonding between the silanol groups with the matrix generating more order molecule i. e more river lines. The SEM micrograph of composite with 5% of MS content (Fig. 6c) shows the ‘sea-island’ structure. The structure attributed to the efficient interaction between MS nanoparticles with the matrix but the nanoparticles have a strong tendency to agglomerate resulting uneven dispersion. The agglomerate is caused by the formation of a silica-silica aggregated structure due to hydrogen bonding.
It is also noted that there is no silica agglomeration formed in unmodified filler (Fig. 6b). This hinted that the lower energy interaction (i.e., hydrogen bonding) between silica and matrix, during sample preparation for SEM analysis (fracturing) the silica were dispersed into the air. This observation was supported by EDX analysis, which shows there is no silica detected in the sample (Fig. 7a). However, the strong interaction (i.e., covalent bonding) of MS filler with composite matrix shows 2.89% of silica remains intact in the composite matrix (Fig. 7b).
This phenomenon suggested that there is no covalent bonding occurred between the PS with the composite matrix but with the addition of MS in the matrix, results in MS to be chemically bonded with the composite matrix. This finding is further supported by the possible mechanism as predicted in Fig. 4.
CONCLUSIONS
From this investigation it can be concluded that the fumed nanosilica particle was successfully modified with epoxy modifier. The addition of modified silica filler in the composite matrix provides covalent interaction resulting better thermal stability, lower degree of shrinkage compared to blank and PS composite matrix but higher tendency to agglomerate and resulting uneven dispersion in the matrix.
ACKNOWLEDGMENTS
The authors gratefully acknowledge financial support from Intel Technology (M) Sdn. Bhd through grant No. 304/PKIMIA/650293/I104.
REFERENCES
- Barthel, H., 1995. Surface interactions of dimethylsiloxy group-modified fumed Silica. Coll. Surf., 101: 217-226.
Direct Link - Bauer, F., H.J. Glasel, E. Hartmann, H. Langguth and R. Hinterwaldner, 2004. Functionalized inorganic/organic nanocomposites are new basic raw materials for adhesives and sealants. Int. J. Adh., 24: 519-522.
CrossRef - Beker, O., L. Varley and G. Simon, 2002. Morphology, thermal relaxations and mechanical properties of layered silicate nanocomposites based upon highfunctionality epoxy resins. Polymers, 43: 4365-4373.
CrossRef - Ho, T. and C. Wang, 2001. Modification of epoxy resin with siloxane containing phenol aralkyl epoxy resin for electronic encapsulation application. Euro. Polym., 37: 267-274.
Direct Link - Kang, S., S. Hong, C.R. Choe, M. Park, S. Rim and J. King, 2001. Preparation and characterization of epoxy composites filled with functionalized nanosilica particles obtained via sol-gel process. Polymorphism, 42: 879-887.
CrossRef - Kickelbick, G., 2003. Concepts for the incorporation of inorganic building blocks into organic polymers on a nanoscale. Prog. Polym. Sci., 28: 83-114.
CrossRef - Liu, Y., C. Hsu, M. Wang and H. Chen, 2003. A novel approach of chemical functionalization on nano-scaled silica particles. Nanotech., 14: 813-819.
Direct Link - Liu, Y., C. Hsu, W. Wei and R. Jeng, 2003. Preparation and thermal properties of epoxy-silica nanocomposites from nanoscale colloidal silica. Polymer, 44: 5159-5167.
CrossRef - Liu, Y., W. Wei, K. Hsu and W. Ho, 2004. Thermal stability of epoxy-silica hybrid materials by thermogravimetric analysis. Thermo. Acta, 412: 139-147.
CrossRef - Ritzenthaler, S., E. Girard-Reydet and J.P. Pascault, 2000. Influence of epoxy hardener on miscibility of blends of poly(methyl methacrylate) and epoxy networks. Polymorphism, 42: 6375-6386.
CrossRef - Salahuddin, N., A. Moet, A. Hiltner and E. Baer, 2002. Nanoscale highly filled epoxy nanocomposite. Euro. Polym., 38: 1477-1482.
CrossRef - Shirono, H., Y. Amano, M. Kawaguchi and T. Kato, 2001. Characteristic of alkyltrimethoxysilane-treated fumed silicas and rheological behavior of fumed silica suspensions in an epoxy resin. J. Coll. Int. Sci., 239: 555-562.
CrossRef - Stesmans, A., K. Clemer and V.V. Afanas'ev, 2005. Electron spin resonance probing of fundamental point defects in nm-sized silica particles. J. Non-Crys. Sol., 351: 1764-1769.
CrossRef - Wang, H., Y. Bai, S. Lui, J. Wu and C.P. Wong, 2002. Combined effect of silica filler and its interface in epoxy resin. Acta. Mater., 50: 4369-4377.
Direct Link - Wu, C.L., M.Q. Zheng, M.Z. Rong and K. Friedrich, 2002. Tensile performance improvement of low nanoparticles filler-polypropylene composites. Comput. Sci. Technol., 62: 1327-1340.
CrossRef - Wu, C.L., M.Q. Zheng, M.Z. Rong and K. Friedrich, 2005. Silica nanoparticle filled polypropylene: Effects of particle surface treatment, matrix ductility and particle spesies on mechanical performance of the composites. Comput. Sci. Technol., 65: 635-645.
CrossRef - Xing, X.S. and R.K.Y. Li, 2004. Wear behavior of epoxy matrix composites filled with uniform sized sub-micron spherical silica particles. Wear, 256: 21-26.
CrossRef