
Iran Sheikhshoaie
Department of Chemistry, Shahid Bahonar University of Kerman, Iran
Samira Saeid-Nia
Department of Chemistry, Shahid Bahonar University of Kerman, Iran
Niaz Monadi
Department of Chemistry, Shahid Bahonar University of Kerman, Iran
Hojatollah Khabazzadeh
Department of Chemistry, Shahid Bahonar University of Kerman, Iran
Journal of Applied Sciences
Year: 2007 | Volume: 7 | Issue: 1 | Page No.: 145-147







ABSTRACT
Two new trident imine compounds 4-nitrophenylazo- N-(2-hydroxy propylamine) saliciliden [L1] and phenylazo- N-(2-hydroxy propylamine) saliciliden [L2] have been synthesized and characterized by some spectroscopy techniques. The electrical dipole moment (μ) and the first hyperpolarizabilty (β) and amount of charge density on all coordination atoms, HOMO energy and LUMO energy of L1 and L2 compounds were calculated by using AM1 Smi-empirical method. The calculation results reveal this π-conjugated systems have a good NLO property in second harmonic generation technique (SHG). The calculated β (NLO property) for L1 compound is 134.55 times of that Urea.
PDF Abstract XML References Citation
How to cite this article
Iran Sheikhshoaie, Samira Saeid-Nia, Niaz Monadi and Hojatollah Khabazzadeh, 2007. Some Quantum Chemical Study about the Second-Order Non-linearity of Two Imino Chromophores Containing Salen Group. Journal of Applied Sciences, 7: 145-147.
DOI: 10.3923/jas.2007.145.147
URL: https://scialert.net/abstract/?doi=jas.2007.145.147
DOI: 10.3923/jas.2007.145.147
URL: https://scialert.net/abstract/?doi=jas.2007.145.147
INTRODUCTION
Materials possessing nonlinear optical (NLO) properties change the propagation characteristics (polarization, phase, frequency, etc.) of the incident light. The molecules with large optical nonlinearities have recently become the focus of most researches in view of their potential applications in various photonic technologies, including all-optical switching (Kanchana et al., 2002) and data processing especially in optical fibers communication and optical computing which makes the maximum use of light characteristics such as parallel and spatial processing capabilities and high speed (Prasad and Williams, 1990).
A molecule with π-electron system possess many attractive nonlinear optical (NLO) characteristics and show enhanced NLO properties. The design of most efficient organic materials for the nonlinear effect is based on molecular unit containing highly delocalized π-electron moieties and extra electron donor and electron acceptor groups on the opposite sides of the molecule at a appropriate positions on the ring to enhance the conjugation. We have reported some compounds with NLO property elsewhere (Sheikhshoaie, 2003; Sheikhshoaie and Mashhadizadeh, 2003, 2005).
Nonlinear optics is currently an active area of research, development of NLO compounds aim at to optimize higher-order polarizabilities at the molecular as well as material levels (Nalwa and Miyata, 1997).
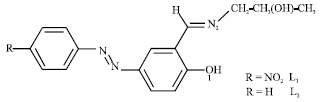 | |
| Fig. 1: | Chemical structure of L1 and L2 imine compounds |
In the present study we synthesized two new tridentate Schiff base ligands L1 and L2 (Fig. 1) and characterized the structures and also we studied their structural properties by using AM1 semi-empirical methods (Kanis et al., 1994). The structure of L1 and L2 Schiff base ligands were shown in Fig. 1.
THEORETICAL CALCULATIONS
Austin model 1 (AM1) (Dewar et al., 1985) is one of the semi-empirical methods and it is a popular method for calculation of the electronic molecular properties such as ground state, geometry, molecular energy and molecular polarizability.
The geometry optimization and hyperpolarazibility calculations were performed using MOPAC 7.0 Program on a Pentium III (550 MHZ processor with 256 MB RAM). The optimized geometry with negative charge density on all coordination sites for L1 and L2 are shown in Fig. 2.
We report βtot (total first hyperpolarizability) for L1 and L2 compounds has been calculated.
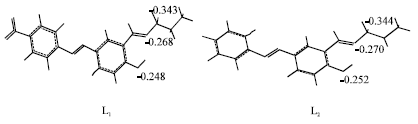 | |
| Fig. 2: | Optimized geometry for L1 and L2 Schiff base compounds by the AM1 semi empirical method |
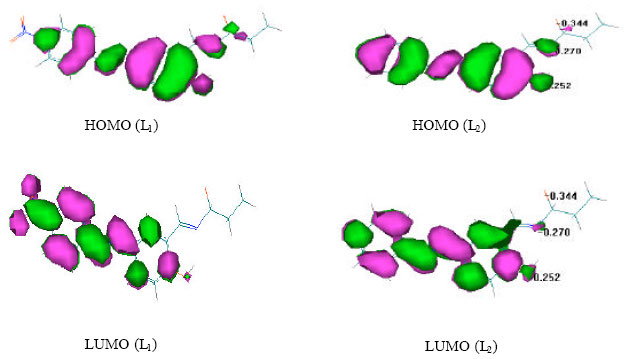 | |
| Fig. 3: | The plot of HOMO and LUMO levels for L1 and L2 compounds |
The components of the first hyperpolarizability can be calculated using the following equation:
| (1) |
Using the x, y and z components, the magnitude of the first hyperpolarizability tensor can be calculated by Eq. 2
| (2) |
The complete equation for calculating the magnitude of first hyperpolarizability from MOPAC 7.0 out put is given as follows8:
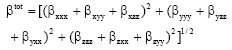 | (3) |
The calculate all the electronic dipole moments and all the first hyperpolarizabilities, the origin of the cartesian coordinate system (x, y, z) = (0, 0, 0) has been chosen at own center of mass of L1 and L2 compounds in Fig. 1. Present calculations indicate that the L1 compound might be the β-interesting material. It is shown that L1 compound has great non-zero μ values (Table 1).
| Table 1: | The AM1 calculated total electric dipole moments μ (Debye) and dipole moment components (μx, μy and μz ) for L1 and L2 imine compounds |
 | |
| Table 2: | The AM1 calculated total first hyperpolarizability βtot for L1 and L2 imine compounds |
Some calculated structural properties like βtot value calculated by AM1 semi-empirical method for L1 and L2 imines compounds are listed in Table 2.
The NLO responses can qualifiedly be understood by examining the energetic of frontier molecular orbitals [the highest molecular orbit (HOMO) and the lowest molecular orbit (LUMO)] of L1 and L2 imine compounds (Table 3). L1 molecule has a NO2 group in para position on phenyl group while L2 molecule has H atom in this position.
| Table 3: | The calculated energy of frontier molecular orbital (eV) for L1 and L2 compounds by AM1 semi-empirical method |
The NO2 group possess strong electron-withdrawing action, when NO2 was attached to the para position of phenyl ring, unshared electron-pair of the system could transfer to NO2 along the conjugation system.
In the other hand the HOMO, largely dictates the source of charge transfer (CT), from the HOMO-LUMO energy calculation by MOPAC 7.0 it can be seen that the HOMO of the L1 molecule is lower than the HOMO level of L2 molecule. According to the HOMO-LUMO differences of above molecules, it can be seen that the HOMO-LUMO gap of L1 molecule is relatively smaller than that of the L2 molecule and shows higher β value than that of the L2 molecule. It is evident that there should be an inverse relationship between HOMO-LUMO gap and the first of hyperpolarizability (Kanis et al., 1994).
Figure 3 shows the electron density in HOMO and LUMO levels for L1 and L2 molecules.
CONCLUSION
| • | L1 and L2 compounds have three atoms as their coordination sites for metal complex formation (O1, N2 and O3, Fig. 1). |
| • | The geometry of L1 and L2 compounds are flat and there is a hydrogen bond between O1 and N2 atom in these structures. |
| • | Our calculations show that L1 and L2 compounds have NLO property, but Table 2 shows that L1 compound is a good candidate for second harmonic generation. |
| • | β (NLO property) calculated for L1 compound is 134.55 times of that Urea (β for Urea is 0.14 x10-30 esu). |
| • | NO2 group has a good role in the NLO property of these compounds (Table 2). |
REFERENCES
- Kanchana, S. and K.N. Nailn, 2002. Nonlinear optical properties of novel fluorenyl derivatives- ab initio quantum chemical calculations. J. Mol. Struc. (Theochem), 617: 169-175.
CrossRef - Kanis, D.R. M.A. Ratner and T.J. Marks, 1994. Design and construction of molecular assembles with large second- order optical nonlinearities. Chem. Rev., 94: 195-242.
Direct Link