
P.I. Antonio Caprai
Institute of Geosciences and Earth Resources, National Research Council of Italy, 56124 Pisa, Italy
Journal of Applied Sciences
Year: 2005 | Volume: 5 | Issue: 1 | Page No.: 85-92







ABSTRACT
Chemical analyses of volcanic, geothermal and low enthalpy fluids are carried on to investigate volatile evolution. The methods used are mainly two: collection of residual gases and total gases. Both are valid, but for different use. In residual gases one can analyze ratio gas/steam, minus and major components but no carbon monoxide because interference with sodium idroxide. In total gas is difficult to analyze minus components but it`s good for carbon monoxide and isotopic composition in carbon dioxide. In both sampling methods the problem is to choose a good way for sampling. This manual shows the methods we consider the best in use. Another problems is to minimize analytical error. It depends from the quality of instrumentation and the quality of methods we use. We describe the methods in use in IGG laboratories that give good results, according with quality of instrumentation, quality of standard and reproducibility. We think that may exist other valid methods to collect and analyze samples. The methods we describe are just a starting point, it depends on the ability of researchers and operators to find methods valid for particular sites. In addition are described preparation of bottles for sampling different kinds of natural manifestations. There are different preparations in residual flasks if we sample volcanic fumaroles or geothermal wells. In addition to this some problems can comes from the pressure inside the bottles. We solve the problem with a particular apparatus self made for working at very low pressure, even under one hundred mbar.
PDF Abstract XML References Citation
How to cite this article
P.I. Antonio Caprai, 2005. Volcanic and Geothermal Gases and Low-enthalpy Natural Manifestations Methods
of Sampling and Analysis by Gas Chromatography. Journal of Applied Sciences, 5: 85-92.
DOI: 10.3923/jas.2005.85.92
URL: https://scialert.net/abstract/?doi=jas.2005.85.92
DOI: 10.3923/jas.2005.85.92
URL: https://scialert.net/abstract/?doi=jas.2005.85.92
INTRODUCTION
In performing analytical determinations[1] geochemists often encounter problems right from the stages of sampling. Methodologies for fluid collection are varied and, at times, rather improvised. Familiarity with the methods normally adopted can prove invaluable to anyone deciding to tackle such an undertaking.
The purpose of this technical manual is to describe some methods of sampling and gas chromatographic and chemical analyses of geothermal and volcanic fluids as well as those originating in other low-enthalpy natural manifestations. A particular attention is given to preparation and choose of metals or glasses for the pipe useful for collecting volcanic fluids. The gases considered are the following: CO2, H2S, SO2, Ar, O2, He, H2, CH4, N2, CO, N2O, H2O, NH3, HCl, HF, C2H6, unsaturated C2, C3H8, C3H6, iso-C4H10, n-C4H10, iso-C4H8, n-C4H8.
Sometimes one’s can have problems in analyze residual gases because of small amount of gas (because of low ratio gas/steam) and/or small pressure inside the bottles.
Materials required for fluid sampling: The term "fluid” is used herein to refer to the vapor and/or all gaseous components issuing from a geothermal or fumarole manifestation. Some essential equipment for the collection procedure are: several meters of heat-resistant silicone tubing of various diameters; a 100 cc plastic gas syringe; a glass and Teflon “T” tap; a suitable number of double-valved glass sample holders for collecting the “total” gas and degassed and vacuumized, single-valved-valved glass sample holder with a suitable amount of 4 N NaOH (normally 50 cc); four funnels - two plastic and two metal, 10 and 25 cm in diameter; two pipes-a metal one (preferably titanium) and a quartz one, perforated at its end; two or three glass traps of about 250 cc; two Dewar flasks, and if possible, some ice dry (alternatives: water and ice, ether or water); alcohol; an indelible marker; various plastic pipe fittings; scissors; screwdriver; pliers; metal clamps; a notebook; pens and pencils.
Preparing the sample holder: When a double-valved glass sample holder (Fig. 1) is used for collecting total gas, a lubricant and sealant grease with the followings characteristics must be used: high adhesion under vacuum with a very low vapor pressure; chemical unreactivity and good lubricating capacity. Moreover, it must be able to maintain these properties at both high and low temperatures for a period of at least six months.
At the laboratory of the I.G.G. Apiezon N or T grease is used. Although the “T” type is more resistant to heat, it does not maintain its desirable properties as long as type “N.”
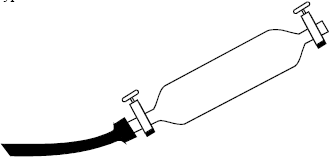 | |
| Fig. 1: | Double-valves glass sample holder fit for total gas sampling |
It is generally preferable to lubricate the taps on site immediately before sampling, especially if reaching the site involves air travel (to avoid alteration of the grease's properties by the extreme cold often found in baggage compartments).
The proper sample holder for collecting residual gases has a valve with Teflon piston (Fig. 2).
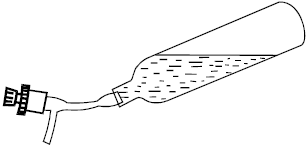 | |
| Fig. 2: | Sample holder for collecting residual gases with a valve with teflon piston |
The sodium hydroxide must not contain CO2 (Labeled: CO2-free NaOH), and the solution must be prepared immediately before being transferred to the sample holder so as to avoid absorption of atmospheric CO2. Small amounts of sodium hydroxide in tablet form are added to preheated water up to the desired concentration; a silicone rubber tube is then introduced into the beaker and the desired quantity of solution aspirated directly into the previously vacuumized sample holder. The sample holder must be vacuumized by interposing a trap with liquid nitrogen or dry ice between the vacuum pump and the sample holder to prevent sodium hydroxide from entering the rotor. The resulting vacuum will correspond to the vapor pressure of 4 N NaOH, that is, about 15 mbar. In case of volcanic fluids, in the sodium solution we added 20 cc of ZnOH 1 N for separation of S= (precipitation as ZnS) from SO3= (dissolved in solution).
Sampling: The main problems in collecting geothermal samples fluid are: contamination with air during sampling or subsequent stages and the non-proportional loss of one or more constituents due to chemical interactions (example, the reaction in humid environments between H2S and O2) or physical ones (e.g., the solubilization of CO2 and H2S in the liquid phase during separation of the gas from the vapor in the sampled fluid).
There are two types of gas samples, one termed total gas, which is generally dry and contains all components soluble in the condensate water, except for SO2 and NH3. The other is called residual gas, where the minor components are found in the gaseous phase together with CO2, H2S and SO2 in the liquid phase, initially made up of 4 N NaOH. This type of sampling can be used to measure the ratio between the gas and the vapor. In case of low ratio of gas/steam there are problems to have the right amount of gas for gaschromatographic analyses. In this case in our opinion is better to have a low pressure with enough volume of gas then viceversa. In case is possible collect residual gases trough a good separator and make a merging with residual gas collect in the right mode.
The procedure for locating the best site to perform the sampling varies according to the type of manifestation in question.
Sampling from natural manifestations below 100°C: Such low-temperature manifestations are often present in water and mud springs. Mud may be found because clay was either present at the source's outlet or deposited through local transport phenomena. Generally, green algae, whose growth is fostered by the possible presence of ammonia, are also found.
When springs are sampled, the site of greatest gas flow should be determined. If such flow is believed to be sufficient to displace the air present in the sample holder, the funnel is inverted and appropriately ballasted, and then placed over the manifestation (often completely immersed in the water). The funnel is then connected to the total-gas sample holder by means of a silicone rubber tube, and the gas made to flow through the sample holder for as long as is necessary to flush it out thoroughly (the volume of gas made to flow must be at least 5 times that of the holder). The flow of gas is controlled through a rubber tube connected to the sample holder's exit valve, which is immersed in a liquid that enables the bubbling of the outflowing gas to be checked visually.
In the event that the flow is insufficient for proper flushing, one possibility is to fill the sample holder with water and wait for the gas bubbles to displace the water while trying to help the gas rise by gently shaking the system and/or pinching and releasing the rubber tube.
When sampling residual gas (Fig. 3), one common problem encountered is due to the fact that, as the NaOH-containing sample holder is vacuumized, a pumping effect may result that acts to pull the water-dissolved gases out of solution. Considering that the solubility in water of each gas is different, and that water is usually saturated in O2 and N2, these two gases are freed in different ratios from those present in air. In fact, it is not uncommon to find an O2 content of 30% or more in such a sample. The sampling must therefore be carried out slowly, trying to minimize the problems linked to this pumping effect and/or to not suck up the river or the spring water as well.
The solubility of oxygen is: 2.33 cm3 100-1 g H2O under normal conditions
The solubility of nitrogen is: 4.89 cm3 100-1 g H2O under normal conditions
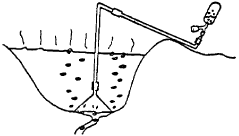 | |
| Fig. 3: | Sampling residual gas from pool with a funnel |
Fumarole sampling: In this case, the most troublesome problems are represented by corrosion of the material, difficult access to the manifestation and the acid fumes that may envelop the field worker[2].
Determining the best site to perform such sampling is made on the basis of the flow rate and/or the maximum temperature found in the zone in question (from 100 to over 800°C). Temperature measurements are performed with a thermocouple. If available, a titanium pipe should be introduced into the fissure; otherwise quartz tubes have been used successfully. It is unadvisable to use iron instruments for collecting the fluid because in the presence of a water condensate, the following reaction takes place:
The production of H2, apart from increasing the concentration of this gas itself, also leads to serious errors in analyzing its isotopic composition.
After having positioned the pipe, it should be sealed to avoid contamination by the surrounding air (the pipe is about 50 cm long). Then, after connecting the rubber tubing, it is advisable to wait for a few minutes, so that the fluid flowing through the tubes has a chance to heat them in order to avoid possible isotopic separation of the vapor. When sampling total gas (Fig. 4) the fluid must be made to flow through a condenser which holds back the vapor. This is for two reasons: the first is that in a dry gas the reaction kinetics between H2S and any O2 is very low (with water acting as catalyst), while the second is that the condensed vapor and CO2 are usually analyzed in the form of their stable isotopes, and the presence of humidity in the sample holder would be an index of a more or less pronounced degree of fractionation of the water collected. Moreover, fractionation of the O2 in the CO2 would be caused by isotopic exchange with the oxygen of the water present in the sample holder.
Proper sampling can generally be achieved by using two traps immersed in a solution of alcohol and dry ice that lower the internal temperature to about –80°C. An alternative refrigerant system that can be used, such as dipping the traps in either ice water or ether. The fluid passes through the traps before picking up the gas. Any vapor present condenses, thus being separated from the gas, which can then flow into the sample holder, on whose outlet a bubbler with a bit of Vaseline oil has been placed in order to check the regularity of flow.
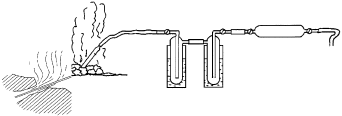 | |
| Fig. 4: | Sampling total gas through two condenser traps immersed in dry ice and alcohol |
Whenever the fluid does not issue forth with sufficient speed, the gas must be forced out with a peristaltic pump or a syringe connected to a "T" tap. Suction must be applied slowly, so as not to introduce air from the exterior.
Regarding residual gas, the sample holder is connected directly to the tube leading from the pipe (Fig. 5). The entry of vapor and the exothermal reaction between the NaOH solution and CO2 causes an increase in temperature that slows the reaction. By cooling and gently shaking the sample holder, the process can be facilitated.
Using this method, all the fluid enters the bulb so that the gas/vapor ratio can be determined, as well as the constituent chemicals.
 | |
| Fig. 5: | Residual gases collected from a fumarole by a pipe in titanium or quartz |
Sampling for geothermal surveys: The collars of wells drilled in geothermal areas are generally fitted with a valve with a drift plug. The fluid discharge is usually very high and the temperatures always under 300°C.
An important factor to be considered is the relative ease of access to the sampling site, and therefore the ability to transport all the necessary equipment.
The total-gas sample holder is to be placed downstream from a water separator (the condensate can be used for analyzing tritium and/or stable isotopes) or two traps immersed in a refrigerant mixture (the condensate is to be used for stable isotope analysis). The separator, however, must be used when the gas/vapor ratio is below 8 mMoles gas / Mole H2O.
The flow through the sample holder (Fig. 6) is controlled by the valve on the collar. Nevertheless, a T-junction must be used for the sake of safety, as well as to keep the system hot; this will discharge any excess pressure laterally.
The tubes downstream from the traps must be kept at ambient temperature, otherwise the vapor will not condense completely, with consequent problems in isotopic fractionation.
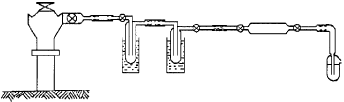 | |
| Fig. 6: | Sampling total gas from geothermal well |
Residual gas sampling is carried out by connecting a heat-resistant rubber tube directly to the well collar. A T-junction (Fig. 7) inserted into the tube will serve to avoid problems similar to those described in the foregoing. In geothermal wells the quantity of vapor is always high; thus, one of the main problems in performing a residual-gas sample is that the volume occupied by the gas may be too low (less than 50 cc) because too much water is present. In such cases, analytically, it is preferable to have a gas under low pressure, but which occupies a volume of at least 100/150 cc. This can be achieved by increasing the concentration of NaOH and introducing half the usual amount (about 20/25 cc), and taking care to halt the sampling as soon as the volume of gas deemed appropriate has been reached.
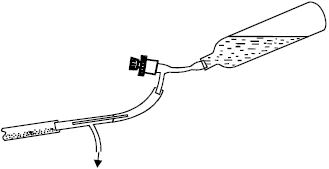 | |
| Fig. 7: | “T” for sampling residual gases from geothermal wells |
It must be kept in mind that high- temperature and pressure fluids are being handled, and it is therefore imperative to take the necessary safety precautions (gloves, visor, helmet, etc.).
Useful suggestions for all types of sampling: In all sampling sessions, a log should be kept, not only of indications of a general nature, but of any problems encountered in order to aid in understanding eventual “anomalies” found in the analytical results. Moreover, it is also generally advantageous if the researcher conducting the analysis is also the same person who has prepared the sample holder and collected the fluids.
Transporting the sample holders: After each sampling, it must be checked that transportation of the samples to the laboratory does not accidentally cause the valve to break or open. The field worker who has performed the fluids collection should take care to apply some adhesive tape to keep the valves from twisting open (particularly the total gas samples because of their arrangement). Sample holders should be wrapped in paper or foam rubber and put in special shock - absorbing containers.
Analysis: Samples analysis is conducted primarily through gas chromatography techniques and, in the case of residual-gas CO2, SO2 and H2S, the classical techniques of volume titration as well. Special sampling and analysis techniques are used for determining ammonia content.
General aspects of gas chromatography: Chromatography is a chemical-physical method of separation based on the equilibrium partition of the components to be separated, which are distributed between two phases: one fixed and the other mobile. The first is made up of a solid or opportunely supported liquid (stationary phase), while the second is represented by a fluid (mobile phase), containing the components to be separated, which percolate through the first.
This method, discovered by the Russian botanist Tswett in 1906, who employed it to separate vegetables components, was introduced into the field of chemistry after some decades as an analytical method for separating various substances. Today it is applied in many areas of research and analysis and has been broadened to include numerous working techniques, all based on the same underlying principle, but differing in their practical application.
The term 'chromatography' comes from the fact that the first separations were performed using compounds that were distinguished on the basis of the various color changes they would undergo during the analysis.
Analytical techniques at the I.G.G.: In the laboratory of the I.G.G. two gas chromatographs are used: a Perkin Elmer model 3920 (1978) with thermoconductivity detector (TCD) and a P. E. model 8500 (1989) with flame ionization detector (FID) and catalyst. The detectors serve to transform the passage of each component of the gaseous mixture making up the eluate into an electric impulse. TCD detectors are non-specific and non-destructive, albeit not very sensitive; FID's, instead, are very sensitive, but destructive (they burn combustible mixtures) and specific for hydrocarbons.
The analytical techniques currently used calls for employing various flow and temperature conditions and packed gas chromatographic columns suitable for the type of analyses being performed each time.
The two gas chromatographs are joined one to the other only by a “centralized variable volume” to which the gas in the sample holder is introduced (Fig. 8). Volume regulation allows using a range of volumes varying from 1 to 25 cc (from 26 to 50 cc considering the fixed volumes represented by the tubes and joints). Thus, it is possible to introduce a quantity of gas at fixed volume, but different pressures through the valves of the gas chromatographs (Fig. 9), thereby varying the absolute amount. The system also includes a pressure gauge that provides pressure readings of the sample holder's interior (in order to correct for expansion of the volume introduced).
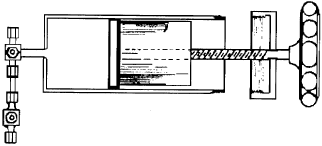 | |
| Fig. 8: | Centralized variable volume |
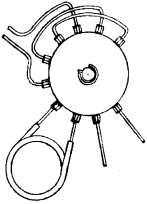 | |
| Fig. 9: | Ten way valve |
The standardization technique used is external: gas mixtures of known concentration are prepared in the laboratory by taking the pure gases and mixing them in the suitable proportions. Mixing occurs in the two-valved sample holder fitted with a porous diaphragm. Such sample holders are filled to normal P with the same gas as that used as carrier, and the components of interest are added with a syringe through the porous diaphragm. The peaks obtained with the standards are used for comparisons with those of the samples in order to determine the percentage concentrations.
The laboratory tests we performed on the I.G.G.'s equipment revealed that the thermoconductivity detectors yield linear responses for all the components analyzed, from the limits of detectability up to a concentration of 100%. Such response, however, depends on not exceeding a gas insertion pressure of over 100 mBar, for which, except for particular cases, quantitative analyses are performed using very high concentrations, if not wholly pure specimens, of standard gas. Regarding the flame ionization detector, variable low concentrations of standards are instead used in order to determine the calibration curve for hydrocarbons and CO.
The Table 1 shows the component groups that can be analyzed under the various conditions. In the case of the I.G.G., two computers, three interfaces and two analysis programs by Perkin Elmer (Omega and Turbochrom) enable the analytical data to be collected, the concentrations calculated and any needed manual adjustments made.
| Table 1: | Illustrates the instrument conditions |
 | |
| TCD: thermoconductivity, FID + Catalyst: flame ionization with zirconium catalyst for reduction of CO, FID: flame ionization | |
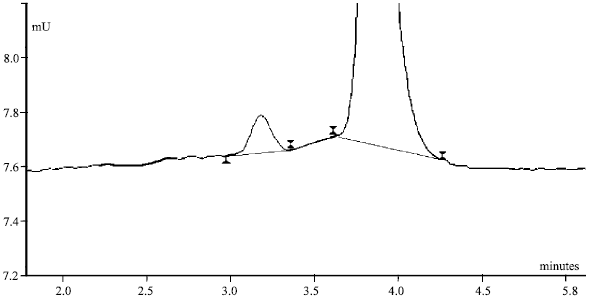 | |
| Fig. 10: | Separation of He (136 ppm) and H2 (1378 ppm); the insertion pressure was 500 mbar. |
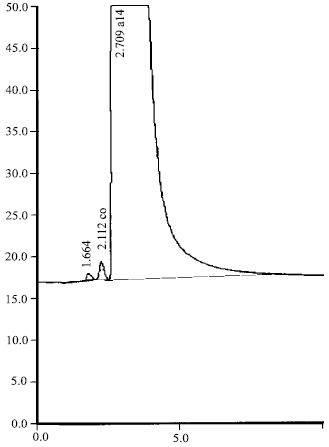 | |
| Fig. 11: | Separation of CO (6.1 ppm) and CH4 (2.81%); the insertion pressure was 500 mbar |
The insertion line of the gas into the columns allows performing more than one analysis with the same sample portion, while at the same time enabling the pressure to be varied. Normally, the insertion pressure is 100 mbar. In order to analyze the components at low concentrations, the pressure can, if necessary, be increased even to over one atmosphere, thereby obtaining an increase in the absolute quantity of gas introduced into column.
Wet analysis: The liquid phase is extracted from the sample holder, its volume measured (V1) and it is maintained in a flask (usually 250 cc) which has been filled with Ar to avoid oxidation of the H2S. A portion of this sample is used for CO2 analysis [ 3 ] .
CO2: The CO2 content is determined in the form of HCO3 - through titration with 0.1000 N HCl.
The reaction is:
At pH 8.25, 99% of all the carbonate present is in the form of bicarbonate.
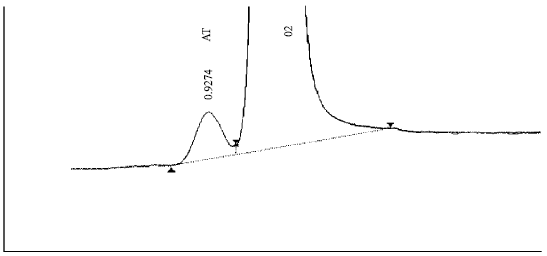 | |
| Fig. 12: | Separation of Ar (093%) and O2 (20.95%) in the concentrations present in air. The working temperature of –20°C requires raising the temperature to 50 °C between one analysis and the next in order to allow all components to be released and avoid phantom peaks in the subsequent run |
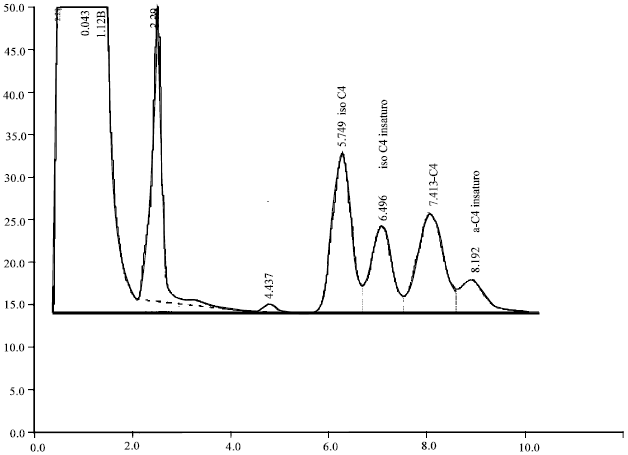 | |
| Fig. 13: | Separation at 100°C of 4-atom carbon compounds in the form of alkenes, as well as the isomers of their respective saturated compounds |
At pH 3.8 all the bicarbonate is converted into CO2.
All other weak acids will add to the results of titration. Therefore, H2S must be removed through sulfate oxidation using H2O2.
Procedure: To a given volume, usually 20 cc, add 2-3 cc of H2O2 and bring the volume to 25 cc (V2). The sample is left to rest overnight and another portion (V3 - generally 5cc) of the oxidized sample transferred to a beaker. The pH is brought to a value of 8.25 using an HCl solution. The titration from pH 8.25 to pH 3.80 with a 0.1000 N solution of HCl yields the quantity of CO2 present.
mM CO2 = V(HCl) * N(HCl) * V2/V3 * V1/V2 |
H2S: H2S is converted to Na2S during the sampling and determined by means of the iodometric method. The sample is added to an acid solution of 0.1000 N I2, whose volume is such that the resulting mixture remains acid even after the addition of the alkaline sample. It is then titrated with a solution of thiosulphate, according to the following reaction[4].
| S= + I v S° + 2I- |
Procedure: A sample volume (V5) is added to 10 cc of 5 N H2SO4, together with a volume (V4) of an I2 solution at known normality N(I2). Titration is then performed with a thiosulphate solution of known N (t), until the starch water indicator changes color.
Calculating the meqs of H2S: meq H2S = ((V4*N(I2) – V(t)*N(t))*V1/V4 (for mM divide the result obtained by 2).
H2S and SO2 in samples of volcanic origin
The supernatant allows the iodometric determination of SO2 according this reaction:
I2 + 2H+ + 2S2O3= v S4O6= + 2HI |
Quantification of H2S is carried on by dissolving ZnS in H2O2 and then using classic ponderal system of precipitation as BaSO4 or ionic chromatographic system for determination of oxidized S= (SO4=).
HCl and HF in samples of volcanic origin
Both components, HCl and HF, normally presents in volcanic fluids are determined by liquid chromatography. Usually them are in large amount, so it is necessary to dilute the main idroxide solution, after oxidation with H2O2.
REFERENCES
- Giggenbach, W.F., D. Tedesco, Y. Sulistiyo, A. Caprai and R. Cioni et al., 2001. Evaluations of results fro the fourth and fifth IAVCEI field workshop on volcanic gases, Vulcano Island, Italy and Java, Indonesia. J. Volcanol. Geothermal Res., 108: 157-172.
Direct Link