
F.M. Mahmoud
Chemistry Department, An-najah N. University, Nablus, P.O Box 7, Palestine
M.A. Al-Nuri
Chemistry Department, An-najah N. University, Nablus, P.O Box 7, Palestine
J.A. Daraghmah
Chemistry Department, An-najah N. University, Nablus, P.O Box 7, Palestine
Journal of Applied Sciences
Year: 2004 | Volume: 4 | Issue: 2 | Page No.: 193-196







ABSTRACT
The rates of cleavage of p-chlorotrimethylsilane were measured in various ROH-DMSO media (R: Me, Et, i-Pr, t-Bu, CF3CH2 -, HOCH2CH2-) and the results were reported. These results have shown that the rates of cleavage decrease as the mole ratio of ROH increases.
PDF Abstract XML References Citation
How to cite this article
F.M. Mahmoud, M.A. Al-Nuri and J.A. Daraghmah, 2004. Solvent Effect on Nucleophilic Cleavage of p-Chlorobenzyltrimethylsilane Using Tetrabutylammonium Fluoride in ROH-DMSO Media. Journal of Applied Sciences, 4: 193-196.
DOI: 10.3923/jas.2004.193.196
URL: https://scialert.net/abstract/?doi=jas.2004.193.196
DOI: 10.3923/jas.2004.193.196
URL: https://scialert.net/abstract/?doi=jas.2004.193.196
INTRODUCTION
The effect of solvent can exert considerable influence on acid and base strength by differential solvation[1]. If a base is more solvated than its conjugate acid, its stability is increased relative to conjugate acid.
It has been shown that solvation by even one molecule can substantially affect the order of basicities[2]. An important aspect of solvent effects is the effect on the orientation of solvent molecules when an acid or base is converted to its conjugate base or acid respectively. For example, an acid RCOOH converted to RCOO- in an aqueous solution. The solvent molecules, by hydrogen bonding, arrange themselves around COO- group in a much orderly fashion than they had arranged around the COOH group. This represents a considerable loss of freedom and a decrease in entropy.
A change from a protic to an aprotic solvent can also affect the acidity or basicity, since there is a difference in solvation of anions by a protic solvation and an aprotic one[3]. The effect can be extreme: in DMF, picric acid is stronger than Hbr,[4] though in water HBr is far stronger. This particular result can be attributed to size. That is, the larger ion is better solvated by DMF than the smaller ion[5]. The ionic strength of the solvent also influences acidity or basicity, since it has an influence on activity coefficients.
Tert-butyldimethylsilyl (TBDMS) ethers of primary, secondary and tertiary alcohols and phenolic TBDMS ethers are desilylated to their corresponding alcohols and phenols, respectively, in DMSO at 80°C[6].
It has been shown that the solvent polarity of DMSO is the highest among several solvents such as, 2,2,2-trifluoroethanol, 2-methyl-2-propanol, 2-propanol, ethanol and methanol[7].
Many studies based on structural, thermodynamical and dynamical properties of pure DMSO and (1:3) DMSO-H2O mixture were performed. It has been found that heavy and slowly moving DMSO molecules are stronger competitors for available donated hydrogen bonds than water molecules[8].
MATERIALS AND METHODS
The starting materials used in the preparation of p-chlorobenzyltrimethylsilane was supplied by Aldrich Chemical Company: p-Chlorobenzyl bromide, chlorotrimethylsilane, tetrabutylammonium fluoride hydrate, calcium chloride, sodium sulphate, magnesium turnings, iodine crystals.
The rate measurements at selected wavelength used to monitor the reactions of p-chlorobenzyltrimethylsilane were carried out using ATI Unicam–UV/ Visible software V-2.11 spectrophotometer. Kinetic measurements were carried out using CPS-240-A-UV/Visible spectrophotometer with thermostatic attachments. The products of cleavage were identified using computerized Fourier Transform Infrared spectrophotometer (Shimadzu FTIR 820 IPC), which has hypride software.
Preparation of p-chlorobenzyltrimethylsilane: The following general method was used. A flame-dried 3-necked round bottomed flask equipped with magnetic stirrer, a double surface reflux –condenser with calcium chloride drying tube and a pressure equalizing dropping funnel, was charged with magnesium turnings. The reaction was initiated by the addition of few drops of neat p-chlorobenzyl bromide and a small crystal of iodine followed by local heating (hair dryer). When the reaction had started the appropriate alkyl halide, diluted with twice its volume of ether (to give an approximately 30% solution) was added at such a rate as to maintain a gentle reflux. After the addition, refluxing was continued for another ½-1 h and then an ethereal solution of p-chlorobenzyltrimethylsilane was added dropwise with stirring. The mixture was heated at reflux for additional 4-8 h, white slurry of magnesium salts usually formed. The mixture was subsequently cooled and then treated with dilute hydrochloric acid (10%) and extracted with ether. The combined ether extracts were washed with water, dried overnight over anhydrous sodium sulphate. The ether was removed by distillation and the residue was distilled, p-chlorobenzyltrimethylsilane was obtained, boiling point 231-232°C, n23D=1.5108
Selection of the appropriate wavelengths: The appropriate wavelengths used to monitor reactions between TBAF and p-chlorobenzyltrimethylsilanes was selected as follows: After addition of a measured concentration of TBAF solution (dissolved in the appropriate medium) to the silane (4-5 X 10-6Kg), the mixture was transferred to a stoppered quartz cell (10 mm path length). The blank solution used in the reference cell was the same as the reaction medium except for the silane. The spectrophotometer was scanned to measure the absorbance in the wavelength range (200-400 X 10-9 m). The plots of wavelength versus absorbance were monitored at intervals. The wavelength at which the absorbance was shifted toward the substituted toluene region was recorded and used later to monitor that reaction.
The wavelength that was obtained and used in the rate studies of p-chlorobenzyltrimethylsilane was 284 X10-9m.
Rate measurements: The technique employed for rate measurments was as follows: 4.2 X 10-6 Kg of the p-chlorobenzyltrimethylsilane was placed in a 5 mL volumetric flask and a measured concentration of tetrabutylammonium fluoride (in the appropriate medium) was added to the mark of the flask after preheating to the required temperature. The mixture was shaken and transferred to the quartz uv cell. The absorption readings were recorded at convenient intervals up to about three half-lives; the infinity readings were taken after ten half-lives.
Isolation of the cleavage product: A solution of 2.4 X 10-3 Kg (12.2 X 10-3 mol) of p-chlorobenzyltrimethylsilane and 4 X10-3 Kg (12.6 X 10-3 mol) of TBAF in 20 mL alcoholic 98% DMSO was refluxed for 8 h and then added to water. The mixture was extracted by ether followed by washing, drying over anhydrous sodium sulphate and fractionation of the ethereal layer gave p-chlorotoluene (60%), boiling point, 162°C, nD20 = 1.5210.
Identification of the cleavage product: The identity of the cleavage product was confirmed by:
UV/Visible spectrophotometer: A 5 X 10-6 Kg of the isolated product was dissolved in 5 mL of alcoholic 98% DMSO and transferred to the stoppered quartz cell (10-2 m path length). The reference cell was filled with alcoholic 98% DMSO. The spectrum obtained was identical with that expected for p-chlorotoluene.
Infrared spectrophotometer: The isolated product was diluted with carbon tetrachloride and the spectrum obtained was identical with that obtained for p-chlorotoluene.
Thin layer chromatography (TLC): TLC was performed for identification of the cleavage product as follows:
A silica gel TLC plate was spotted with p-chlorotoluene and the cleavage product using [petroleum ether: ethyl acetate (2:1) as the eluant]. The two compounds gave the same Rf value.
RESULTS AND DISCUSSION
The rates of cleavage of p-chlorotrimethylsilane depend on both the concentrations of the silane and the nucleophile. The observed rate constants for cleavage of the p-chlorotrimethylsilane were obtained from the slopes of linear plots of ln (At-A8) versus time (Table 1)
The specific rate constants, ks (observed rate constant/TBAF concentration), increased progressively as R is changed from Me to t-Bu (Table 1). The relative ks values being Me, 1; Et, 1.78; i-Pr, 3.5; t-Bu, 16.1.
This sequence can be accounted for in terms of increasing the basicity of the medium as R is changed from Me to t-Bu. The basicity decreases in the following order t- Bu > i-Pr > Et > Me[9].
The extent of the solvation of fluoride and alkoxide anions plays a significant role; especially it is more solvated than the transition state. The more polar the solvent the more solvated the anions and so the more the decrease in the rate of cleavage of carbon-silicon bond.
The polarities of these solvents decrease in the following order: MeOH > EtOH > i-PrOH > t-BuOH.
The bulkiness of the solvent may have a significant effect on the solvation of the anions, as we have seen from the higher rates of cleavage in t-butanol compared to that in methanol.
The following mechanism of this cleavage has been suggested previously by Mahmoud et al. [10].
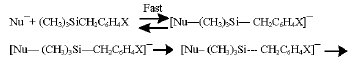 | (1) |
| (2) |
| (3) |
Where Nu¯ represents the fluoride and alkoxide anions but mainly fluoride anion. Since the alkoxide anion is produced in step (3) of the reaction.
The cleavage of carbon-silicon bond was facilated by two factors: i) the long silicon-carbon bonds permit nucleophilic attack at silicon center and ii) the vacant d-orbitals of silicon permit nucleophilic attack via geometric approaches not permitted by the bonding and anti-bonding orbitals of carbon[11,12].
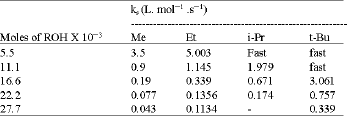
Variation of proportions of ROH in ROH-DMSO media (R: Me, Et, i-Pr, tBu): The rates of cleavage of p-ClC6H4CH2SiMe3 were measured in various proportions of ROH in ROH-DMSO media.
The results in Table 2 show that the rates of cleavage decreases as the number of moles of ROH increases. These results can be attributed to the increasing polarity of the medium as the proportion of ROH increases and so the nucleophile becomes more solvated and its nucleophilic activity decreases (hydrogen bonding to the anion reduces its activity) and so the rate of cleavage decreases[13].
The marked effect, of variation of proportions of ROH on the rates of cleavage, was observed when the number of moles of ROH are changed from 5.5x10-3 to 11x 10-3 , that the rate is decreased by 5.8 for isopropanol , 4.4 for ethanol and 3.9 for methanol, i.e. the rates are more influenced by increasing the number of moles of the more basic medium , t-Bu > i-Pr > Et> Me. Since as mentioned earlier the basicity of the medium increases as the mole ratio of DMSO increases[14].
Reaction in 1, 2-ethanediol – DMSO and CF3CH2OH-DMSO media: The rate of cleavage of p-ClC6H4CH2SiMe3 was measured under the same reaction conditions used for cleavage of the same compound in ROH-DMSO medium expect that 1, 2-ethanediol (1.1x 10-2 mol) was used instead of ROH. The specific rate constant was 4.3x 10-2 L.mol-1 s-1.
The lower rate of cleavage in this medium compared to other ROH-DMSO media reflects its lower basicity and hence higher solvation of the nucleophile, especially, 1, 2- ethanediol has two hydroxyl groups, which are used in hydrogen bonding.
| Table 1: | Specific Rate Constants of Cleavage of p-ClC6H4CH2SiMe3 by TBAF [0.01M] Using Fixed Mole Ratio of 0.01 ROH of 0.01 mol in (ROH-DMSO) mixtures at 30°C |
 | |
| Table 2: | Specific Rate Constants ks of Cleavage of p-ClC6H4CH2SiMe3 by TBAF [0.01 M] in Various Concentrations of ROH in ROH-DMSO Media at 30°C |
 | |
| Table 3: | Specific Rate Constants ks of Cleavage of p-ClC6H4CH2SiMe3 by TBAF [0.01 M] in Various Concentrations of ROH in ROH-DMSO Media at 30°C |
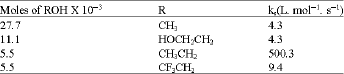 | |
Table 3 has shown a comparison of the specific rate constant obtained in 1,2-ethanediol-DMSO medium with that obtained in MeOH-DMSO. It is found that the rate of cleavage in 1.1x 10-2 mol 1,2-ethanediol parallels of that measured in 2.77x 10-2 mol methanol. This means that the retarding effect of 1.1x 10-2 mol 1, 2-ethanediol on cleavage equal to 2.77x 10-2 mol methanol. Since 1, 2-ethanediol contains two OH groups and since its molecular weight is nearly double that of methanol, the effect of 1.11x 10-2 mole will be similar to the effect of 2.77x 10-2 mol of methanol.
From Table 3, the rate of cleavage of p-ClC6H4CH2SiMe3 by 0.01M TBAF was measured in CF3CH2OH-DMSO medium (5.5x 10-3 mol CF3CH2OH-DMSO) at 30°C with ks value of 9.4x10-2 L.mol-1 s-1 . A comparison of specific rate constants in this medium with those in other media shows that the rate is very low. This result can be attributed to the lower basicity and higher polarity of CF3CH2OH, so that the fluoride anion is strongly solvated.
REFERENCES
- Taft, R. and F. Bodwell, 1988. Structural and solvent effects evaluated from acidities measured in dimethyl sulfoxide and in the gas phase. Acc. Chem. Res., 21: 463-469.
CrossRef - Zhengkun, Y. and J. Verkade, 2000. P(MeNCH2CH2)3N: An efficient catalyst for the desilylation of tert-butyldimethylsilyl ethers. J. Org. Chem., 65: 2065-2068.
CrossRef - Vishnyakov, A., A. Lyubartsev and P. Laaksonen, 2001. Molecular dynamics simulations of dimethyl sulfoxide and dimethyl sulfoxide-water mixture. J. Phys. Chem., 105: 1702-1710.
Direct Link