
R. Radha
Department of Computer Science, S.D.N.B. Vaishnav College For Women, Chromepet, Chennai-44, India
S. Jayalakshmi
Department of Computer Science, S.D.N.B. Vaishnav College For Women, Chromepet, Chennai-44, India
S.P. Rajagopalan
School of Computer Science and Engineering, M.G.R. University, Chennai, India
Information Technology Journal
Year: 2008 | Volume: 7 | Issue: 1 | Page No.: 175-179







ABSTRACT
In this study we show that the gene-expression profiles based on microarray analysis can be used to predict the genes which are responsible for death of the patient in early-stage B-cell Lymphoma. This study presents a new computational method for this prediction and use an informative subset of samples to identify genes whose expression is related to the problem under study. A reduction technique integrating global normalization, fuzzy membership generation, Pearson`s correlation method are used to identify genes which are related to non survival. The early identification of the genes causing B-cell Lymphoma that leads to death will benefit patients in the sense that the patients can be provided proper therapy to increase their life span. The validity of the results are established with the help of statistical methods.
PDF Abstract XML References Citation
How to cite this article
R. Radha, S. Jayalakshmi and S.P. Rajagopalan, 2008. Analysis of Gene Expression Profile to Select Patient Samples for Outcome Prediction. Information Technology Journal, 7: 175-179.
DOI: 10.3923/itj.2008.175.179
URL: https://scialert.net/abstract/?doi=itj.2008.175.179
DOI: 10.3923/itj.2008.175.179
URL: https://scialert.net/abstract/?doi=itj.2008.175.179
INTRODUCTION
Microarray technology permits monitoring of the expression levels of thousands of genes simultaneously. A few previous studies have shown promising results for outcome prediction using gene expression profiles for certain diseases (Rosenwald, 2002; Beer, 2002; Van de Vijver, 2002; LeBlanc, 2003; Fayyad and Irani, 1993). This kind of analysis provides techniques to predict disease progression and clinical outcome at the molecular level. It also identifies genes which are responsible for non-survival of patients. Carefully verifying and understanding these genes would lead to innovative therapies and may also generate opportunities for drug discovery. Various approaches have recently been used on outcome prediction using gene expression profiles. It has been shown that specific patterns of gene expression occur during different biological states such as cell development and during normal physiological responses in tissues and cells.
Generally speaking the expression of genes provides a measure of how active a specific gene is under certain biochemical conditions. This level of expression is related to the relative concentration of messenger RNA (mRNA) which encodes the gene under considerations. The generation of quantitative expression patterns of thousands of genes can be achieved by using techniques based on complementary DNA (cDNA) microarrays (Alizadeh et al., 2000). Various approaches have recently been used on outcome prediction using gene expression profiles. In the Cox proportional hazard regression method (Cox, 1972; Lunn and McNeil, 1995) genes most related to survival are first identified by a univariate Cox analysis and a risk score is then defined as a linear weighted combination of the expression values of the identified genes (Beer, 2002; Rosenwald, 2002). In Ando and Katayama (2002), gene expression profiles are fed to a Fuzzy Neural Network (FNN) system to predict survival of patients. They first predict the outcome of each patient using one gene at one time. Then they rank each gene by their accuracy. Next, one by one, they use the ten highest ranked genes and the selected partner genes for prediction. Finally, the formed ten FNN models using combinatorial genes are optimized by the back-propagation method. In Park et al. (2002), gene expression data are linked to patient survival times using the partial least squares regression technique which is a compromise between principal component analysis and ordinary least squares regression. In Shipp et al. (2002), the weighted voting algorithm is used to identify cured vs fatal for outcome of diffuse large B-cell Lymphoma. The algorithm calculates the weighted combination of selected informative marker genes to make a class distinction. In LeBlanc et al. (2003) they develop a gene index method to investigate genes that jointly relate to patient outcome and to a specific reference gene of interest. The study by Liu et al. (2004) presented a methodology for the identification and filtration of genes.
In this study, we present a new computational method for outcome prediction using gene expression profiles. In the first step, we carefully form samples by selecting only short-term survivors who died within a short period and long-term survivors who were still alive after a relevant long follow-up time. This idea is motivated by our belief that short-term and long-term survivors are more informative and reliable (than those cases in between) for building and understanding the relationship between genes and patient outcome.
In the first step we have divided the DLBCL (Diffuse Large B-Cell Lymphoma) gene expressed database into survival and non survival patient genes. For this separation we have taken the clinical database to identify the patients who died within the prescribed number of years i.e those who were marked as short-term survivors. Then we have selected over regulated genes from the DLBCL gene expressed dataset and then normalized the data set. The dataset is filtered to reduce the dimensionality by applying fuzzy technique and Pearson’s correlation. The filtered data is then analyzed using analysis of variance to find out the homogeneity of genes.
CLASSIFICATION OF DIFFUSE LARGE B-CELL LYMPHOMA USING GENE EXPRESSION DATA
A recent effort to understand how genes contribute to disease, an approach to the discovery of sub-classes of diffuse large B-cell Lymphoma (DLBCL) by using expression analysis is done (Alizadeh et al., 2000). B-lymphocytes are a fundamental component of the body immune system. DLBCL is a malignancy of mature B-lymphocytes, with a high annual incidence in western countries. It has been shown that the discovery of sub-classes in DLBCL has not been successful by relying exclusively on morphological features (Alizadeh et al., 2000). Accordingly Alizadeh et al. (2000) demonstrates that the molecular profile of a tumor obtained from cDNA microarrays can indeed be interpreted as a robust and clear picture of the tumors biology. Additionally, they demonstrate the existence of two molecularly distinct forms of DLBCL that indicate different stages of B-cell differentiation. The prediction and the analysis part of the this paper are tested on the DLBCL domain. They are based on the analysis of the microarray data generated by Alizadeh et al. (2000). The full data and experimental methods are available on the World-Wide Web site of Alizadeh et al. (2000, http://llmpp.nih.gov/lymphoma).
SELECTION OF INFORMATIVE TRAINING SAMPLES
We begin with a new idea to select an informative subset of training samples and then we describe how to identify relevant genes based on these samples. Let D be a training data set for a survival study, that D usually contains two classes of samples Ddied and Dalive. Here, Ddied is the set of patients who died within x (x years/ x months) and Dalive the set of patients who were still alive after x (x years/ x months). A widely used value for x is 5 (years). An informative subset of training samples for D is then the union of a subset of Ddied and a subset of Dalive. The subset of Ddied are those patients who died in a short period (e.g., 1 year) -named as short-term survivors; while the subset of Dalive are those patients who were alive after a long period (e.g., 8 years) named as long-term survivors. Note that long-term survivors may include those patients who died after the specified long period. The short-term and long-term survivors are called extreme cases. This idea emphasizes that the extreme cases play more important roles for survival prediction than those in the middle status i.e., we do not expect reliable prediction could come out from analyzing alive patients whose available follow-up time is short. This idea also helps the identification of those genes that are closely relevant to non survival.
For an experimental sample T if its follow up time is F(T) and status at the end of follow up time is S(T) then:
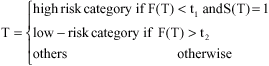 |
Where:
| S(T) | = | 1 stands for an unfavorable outcome. |
| S(T) | = | 0 stands for a favorable outcome. |
| t1 and t2 | = | Thresholds of non survival time for selecting short term and long term non survivors. |
The two thresholds vary from disease to disease or from dataset to dataset. The basic guideline for selection of t1 and t2 is that the informative subset should be reduced to one third or one half of the total available samples.
METHODS FOR RECOGNITION OF RELEVANT GENES
Normalization: For cDNA microarrays, the purpose of dye normalization is to balance the fluorescence intensities of the two dyes (green Cy3 and red Cy5 dye) as well as to allow the comparison of expression levels across experiments (slides) (Yang et al., 2002). Dye bias can be most obviously seen in an experiment where two identical mRNA samples are labeled with different dyes and subsequently hybridized to the same slide. In this situation, it is rare to have the dye intensities equal on average and often the intensities are higher for the green dye. This bias can stem from a variety of factors including physical properties of the dyes (heat and light sensitivity, relative half-life), efficiency of dye incorporation, experimental variability in probe coupling and processing procedures and scanner settings at the data collection step. In this case, a constant adjustment is commonly used to force the distribution of the log-ratios. The purpose is to identify a subset of sharp discriminating features. After narrowing down, the remaining features become sharply discriminating. This systematic method usually selects a reduced percentage of original features. Application of microarrays range from the study of gene expression in yeast under different environmental stress conditions to the comparison of gene expression profiles for tumors from cancer patients. By comparing gene expression in normal and diseased cells microarrays may be used to identify diseased genes and targets for therapeutic drugs. The relative gene expression levels are measured by log ratios from replicate experiments may have different spreads due to difference in experimental conditions. Some scale adjustment is then required so that relative expression levels from one particular experiment do not dominate the average relative expression levels across replicate experiments. All genes on the array are used for normalization. Normalization balances red and green intensities. Imbalances can be caused by different incorporation of dyes, different amounts of mRNA and different scanning parameters. In practice, we usually need to increase the red intensity a bit to balance the green. For this purpose we normalize the dataset. Global methods assume that the red and green intensities are related by a constant factor. That is, R = k • G and in practice, the center of the distribution of log-ratios is shifted to zero:
log2 R/G→log2 R/G-c = log2 R/(kG) |
A common choice for the location parameter log2 k is the median or mean of the log-intensity ratios for a particular gene set (Yang et al., 2002). There are many normalization methods exists. In this paper we have used the global normalization technique.
Discretization by fuzzy membership generation: Uncertainty in ratio measurements are generally greater at lower intensities. Fuzzy logic can efficiently manage the uncertainty and the vagueness of the expression levels. It allows to evaluate the truth-value of a proposition such as g is over expressed under condition i. In binary logic, the previous proposition may be true or false. In fuzzy logics, ![]() is associated a characteristic function
is associated a characteristic function ![]() that defines its membership of the over expressed class.
that defines its membership of the over expressed class.
In most cases, the resulting intervals are not too meaningful and are hard to understand. Chen and Chen (2002) uses fuzzy set to soften partition boundary of the domains and presents the concept of fuzzy association rules, but it does not present partition algorithm that can embody the actual distribution of the data and does not present the mining algorithm for fuzzy association rules which fits for large database. Discretization by fuzzy membership generation (Bernadet, 2000) to all the genes partitions the value range of a numeric feature such that each of the resulting intervals contain the same class of samples, as many as possible. Here the normalized data are further normalized by using the fuzzy triangular membership generation such that the data lies between (0,1), i.e., the degree of membership of the intensity ratio of the particular gene in different patients are expressed.
Correlation: Pearson’s correlation is used to measure the correlation between two features. It is a widely used measure in the analysis of gene expression data. In this paper correlation near one are taken in to consideration since they express the same behavior and a threshold is set in advance for r and subgroups are formed based on a given group of features. In this we set a threshold rc for r in advance, so that r(X,Y)>rc, then features X and Y are correlated. We set rc = 0.9 in this paper. Finally representatives from all the subgroups are taken for non survival risk estimation and outcome prediction. Finally filtering process discards many unrelated genes and only keeps a small number of informative representatives. To find out the homogeneity of genes and patients, the analysis of variance technique (Panneerselvam, 2005) is used and conclusions are derived. Feature sub grouping is done by correlation test where rc is the Pearson’s correlation coefficient and a threshold is taken which is ≥0.9
Figure 1 shows the pseudo code of correlation method used and Fig. 2 shows the algorithm of overall process.
 | |
| Fig. 1: | Pearsons correlation algorithm adopted |
 | |
| Fig. 2: | Overall process |
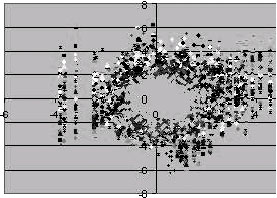 | |
| Fig. 3: | Representation of non-normalized data |
RESULTS
Survival for diffuse large B-cell Lymphoma (DLBCL) patients was previously studied by (Rosenwald et al., 2002) using gene expression profiling and Cox proportional-hazards model. For our study, we select only two extreme cases-long-term and short-term survivors and we have taken non survival from DLBCL data sets.
The original and normalized data is shown in Fig. 3-5.
 | |
| Fig. 4: | Normalized data (global normalization) |
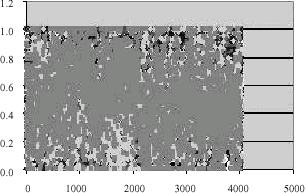 | |
| Fig. 5: | Using fuzzy membership |
Figure 3 shows how the data is distributed. Then after using global normalization we can see how the data is normalized in Fig. 4. Finally applying fuzzy logic the data is brought in such a way that all are centered within range [0, 1]. Now after using the steps 4 and 5 of Fig. 2 we are able to filter the genes from 4026 to 1417 genes. Then using step 6 we are able to reduce to 527 genes which are again reduced by applying Pearson correlation method which is shown in Fig. 1 . In Liu et al. (2004) the filtering process is done drastically much in the entropy measure itself. There are chances that it will leave the most of the genes which are having higher degree of causing disease. In our process since the generation of fuzzy membership values automatically takes into account the overlapping regions, we are able to identify not only the high intensity genes but also genes whose intensity is nearer to the high intensity genes.
In the step of gene identification, built on fuzzy and statistical knowledge, our filtering process discards many unrelated genes and only keeps a small number of informative representatives. Most importantly these genes play a vital role in performance. The dataset on DLBCL (Alizadeh et al., 2000) contains 4026 genes and 47 samples.
| Table 1: | The filtering progress steps |
 | |
The data is normalized using the global normalization explained earlier and by applying the fuzzy membership generation to find the degree of membership of genes on the samples has reduced the data by 35% and a scoring function is used to further reduce the data. Then correlation is used to identify the final set of genes which are responsible for non-survival. The analysis of variance technique used shows that the final set of genes selected are similar in characteristics and are responsible for causing the disease and patients affected by these genes are in the different critical state under non survival group. Table 1 shows the methods we have used to filter the genes.
CONCLUSIONS
In DLBCL study, we first considered all the 4026 genes. The gene filtering technique done by fuzzy membership generation, scoring and correlation leads the final reduction to 77 genes. If we are able to identify these genes among the survival group at an earlier stage, appropriate medical treatment can be given to increase their life span. The future study may be concerned with the identification of a very small number of vital genes which are responsible to predict the outcome from a sample for any related problem under study.
REFERENCES
- Alizadeh, A.A., M.B. Eisen, R.E. Davis, C. Ma and I.S. Lossos et al., 2000. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature, 403: 503-511.
CrossRefDirect Link - Ando, T. and M. Katayama, 2002. Selection of causal gene sets from transcriptional profiling by FNN modeling and prediction of lymphoma outcome. Genome Inform., 13: 278-279.
Direct Link - Beer, D.G., L.R.K. Sharon, C.C. Huang, T.J. Giordano and A.M. Levin et al., 2002. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat. Med., 8: 816-823.
CrossRefDirect Link - Chen, S.M. and Y.C. Chen, 2002. Automatically constructing membership functions and generating fuzzy rules using genetic algorithms. Cybernetics Syst. Int. J., 33: 841-862.
CrossRef - Cox, D.R., 1972. Regression models and life-tables. J. Royal Stat. Soc. Ser. B, B34: 187-220.
Direct Link - Fayyad, U. and K. Irani, 1993. Multi-interval discretization of continuous-valued attributes for classification learning. Int. Joint Conf. Artificial Intel., 13: 1022-1029.
Direct Link - LeBlanc, M. et al., 2003. Directed indices for exploring gene expression data. Bioinformatics, 19: 686-693.
Direct Link - Liu, H., J. Li and L. Wong, 2004. Selection of patient samples and genes for outcome prediction. Proceedings of the Computational Systems Bioinformatics Conference, August 16-19, 2004, IEEE Computer Society, Washington, D.C., USA., pp: 382-392.
CrossRef - Lunn, M. and D.R. McNeil, 1995. Applying Cox regression to competing risks. Biometrics, 51: 524-532.
PubMedDirect Link - Maurice B., 2000. Basis of a fuzzy knowledge discovery system. Proceedings of the 4th European Conference on Principles of Data Mining and Knowledge Discovery, September 13-16, 2000, Springer-Verlag, pp: 24-33.
Direct Link - Park, P.J., L. Tian and S. Kohane, 2002. Linking gene expression data with patient survival times using partial least squares. Bioinformatics, 18: S120-S127.
PubMedDirect Link - Rosenwald, A. et al., 2002. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. New England J. Med., 346: 1937-1947.
Direct Link - Shipp, M.A. et al., 2002. Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat. Med., 8: 68-74.
Direct Link - Van de Vijver, M.J., Y.D.He, L.J. van't Veer, H. Dai and A.A. Hart et al., 2002. A gene-expression signature as a predictor of survival in breast cancer. New England J. Med., 347: 1999-2009.
PubMedDirect Link - Yang, Y.H., S. Dudoit, P. Luu, D.M. Lin, V. Peng, J. Ngai and T.P. Speed, 2002. Normalization for cDNA microarray data: A robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res., 30: e15-e15.
PubMedDirect Link