
Saad Osman Abdalla Subair
College of Computing, Al Ghurair University, Dubai, United Arab Emirates
Safaai Deris
School of Graduates Studies, University Teknologi Malaysia, UTM Skudai, Johor, Malaysia
Mohd Saberi Mohamad
School of Graduates Studies, University Teknologi Malaysia, UTM Skudai, Johor, Malaysia
Information Technology Journal
Year: 2005 | Volume: 4 | Issue: 4 | Page No.: 433-438







ABSTRACT
Advances in molecular biology in the last few decades and the availability of equipment in this field lead to the rapid sequencing of considerable genomes of several species. These large genome sequencing projects generate huge number of protein sequences in their primary structures that are difficult for conventional molecular biology laboratory techniques like X-ray crystallography and NMR to determine their corresponding 3D structures. Protein secondary structure prediction is a fundamental step in determining the 3D structure of a protein. In this study a new method for predicting protein secondary structure from amino acid sequences has been proposed and implemented. The prediction method was analyzed together with other five well known prediction methods in this domain to allow easy comparison and clear conclusions. Cuff and Barton 513 protein data set was used in training and testing the prediction methods under the same hardware, platforms and environments. The newly developed method utilizes the knowledge of the GORV information theory and the power of the neural networks to classify a novel protein sequence in one of its three secondary structures classes. The newly developed method (NN-GORV) was rigorously tested together with the other methods and observed outperformed the GOR-V methods by 7.4% Q3 and the neural networks method (NN-II) by 5.6% Q3 accuracy. The Mathews Correlation Coefficients (MCC) showed that NN-GORV secondary structure predicted states are strongly related to the observed secondary structure states.
PDF Abstract XML References Citation
How to cite this article
Saad Osman Abdalla Subair, Safaai Deris and Mohd Saberi Mohamad, 2005. A Hybrid Classifier for Protein Secondary Structure Prediction. Information Technology Journal, 4: 433-438.
DOI: 10.3923/itj.2005.433.438
URL: https://scialert.net/abstract/?doi=itj.2005.433.438
DOI: 10.3923/itj.2005.433.438
URL: https://scialert.net/abstract/?doi=itj.2005.433.438
INTRODUCTION
Advances in molecular biology in the last few decades and the availability of equipment in this field lead to the rapid sequencing of considerable genomes of several species. In fact, to date, several bacterial genomes, as well as those of some simple eukaryotic organisms have been completely sequenced. The Human Genome Project (HGP) is almost completely sequenced with a rough draft announced in the year 2000. These large genome sequencing projects generate huge number of protein sequences in their primary structures that are difficult for conventional molecular biology laboratory techniques like X-ray crystallography and NMR to determine their corresponding 3D structures[1]. Proteins are series of amino acids known as polymers linked together into contiguous chains[2].Protein has three main structures: Primary structure: Which is essentially the linear amino acid sequence. Secondary structure: Which are α helices, β sheets and coils which are formed when the sequences of primary structures tend to arrange themselves into regular conformations[3]. Tertiary or 3D structure: Where secondary structure elements are packed against each other in a stable configuration. One of the main approaches of predicting protein structures from sequence alone is based on data sets of known protein structures and sequences. This approach attempts to find common features in these data sets which can be generalized to provide structural models of other proteins.
The GOR method was first proposed by Garnier et al.[4]. The GOR method is based on the information theory and naive statistics[5]. The mostly known GOR-IV version uses all possible pair frequencies within a window of 17 amino acid residues with a cross-validation on a data base of 267 proteins[6]. The GOR-IV program output gives the probability values for each secondary structure at each amino acid position. The GOR method is well suited for programming and has been a standard method for many years.
Artificial neural networks have great opportunities in the prediction of proteins secondary structures[7]. Since the neural network can be trained to map specific input signals or patterns to a desired output, information from the central amino acid of each input value can be modified by a weighting factor, grouped together then sent to a second layer known as the hidden layer(s) where, the signal is clustered into an appropriate class[8,9]. Feedforward neural networks are powerful tools. They have the ability to learn from examples and they are extremely robust, or fault tolerant. They are usually used and trained to solve the protein secondary structure prediction problems[8].
MATERIALS AND METHODS
NN-GORV-I prediction method depends on the statistical assumption that combining relevant information in different prediction or classification methods will possibly increase the prediction accuracy of the combined method[10]. There is no existing method up to date, combining GORV with neural networks.
The Stuttgart University SNNS neural network simulator program (http://www-ra.informatik.uni-tuebingen.de/SNNS/) is used in this experimental study. SNNS for UNIX X Windows is used to generate many prototypes of neural networks. The snns2c program is used to convert the simulated networks into ANSI C functions codes that are included in the main C program. In this study, GOR-V which is based on the information theory and neural networks which is based on the work of many researchers in the area of protein secondary structure that is sparked by Quian and Sejnowski[9] and then refined by several other researchers[11-13], are combined to achieve a better prediction method.
Conventional GOR methods used windows of 17 residues. This indicates that for a given residue R, eight immediate nearest neighbouring residues on each side are analyzed. If R is considered as R0, then R+8 and R–8 are the immediate neighbouring residues. The information theory allows the information function of a complex event to be decomposed into the sum of information of simpler events, which can be written as:
 | (1) |
Where, how much information difference is calculated as:
| (2) |
Where, n-S are the confirmations that are not S, i.e if S is happened to be E then n-S is the others two states H and C.
In this experiment, the improvements to the original GOR algorithms are implemented following the suggestions of Kloczkowski et al.[14] with considerable modifications.
The data base is composed of 480 proteins compared to the previous GOR database of 267 proteins. The use of this database allows an objective and unbiased calculation of the accuracy of the prediction. The latest version of the GOR-IV algorithm used a window with a fixed width of 17 residues as explained earlier. A resizable window for the GOR-V algorithm is used in this study according to, the length of the sequence as follows:
| • | Sequences 25 residues or shorter length, a sliding window size of 7 residues is used. |
| • | Sequences greater than 25 and less than or equal to 50 residues length, a sliding window of 9 residues is used. |
| • | Sequences greater than 50 residues long and less than 100 residues, a sliding window of 11 residues is used. |
| • | Sequences greater than 100 residues long and less than 200 residues, a sliding window of 13 residues is used. |
| • | Sequences greater than 200 residues long, a window size of 17 residues is used. |
The original GOR algorithms had a tendency to over-predict the coil state (C). The coil state is adjusted that it will be selected as the predicted state only if the calculated probability of the coil conformation is greater than the probability of the other states by (0.15 for E and 0.075 for H). This approach is known as decision constant or adjustable weights and had been applied successfully in PSIPRED algorithm.
PSIBLAST multiple sequence alignments for each protein sequence in the database had been used in this experiment PSIBLAST program is implemented using the nr database with default parameters. The alignments produced by PSIBLAST that are too similar to the query sequence are removed using trimmer program. A detailed representation for the NN-GORV-I prediction method is shown in Fig. 1.
RESULTS AND DISCUSSION
Figure 2 shows that most proteins of the 480 proteins scored a Q3 of above 50%. About 180 proteins scored a Q3 of 80% while above 100 proteins scored a Q3 accuracy of 70% and just below 100 proteins scored an accuracy of 90%. However, few proteins which are less than 10 scored a Q3 of 100% accuracy.
Figure 3 represents a histogram that elucidates the performance of the six prediction methods.
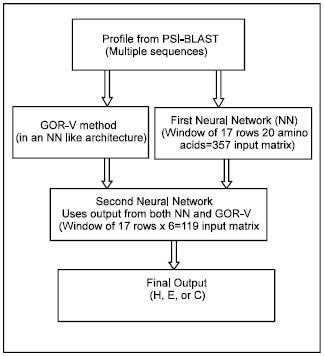 | |
| Fig. 1: | A general model for the newly developed protein secondary structure prediction method |
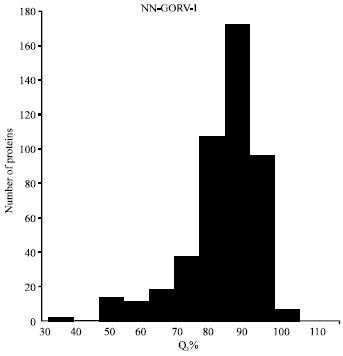 | |
| Fig. 2: | The performance of the NN-GORV-I prediction method with respect to Q3 prediction measures |
It shows the six classifiers Q3 accuracy from the 50% level and above. Based on the nature of the composition of protein secondary structure, it is worth mentioning that prediction accuracy of 50% is worst than random guess[15,16].
Figure 3 shows that the NN-I method predicted about 30 proteins at the level between 50-55% and the PROF and NN-II methods predicted below 20 proteins for each respective level.
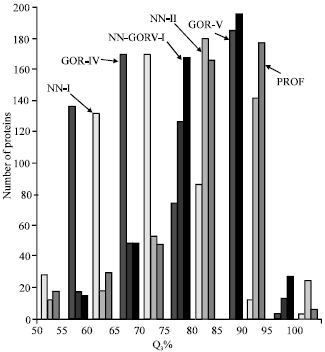 | |
| Fig. 3: | Histogram showing the Q3 performance of the six prediction methods |
This illustrates that these classifiers predict a considerable number of proteins at this low level of 50-55%.
NN-I and GOR-IV methods predict around 120 proteins each at the level of 55-65%. The rest of the prediction methods predicted less than 20 proteins each, except the PROF which predicted about 30 proteins at the 55-65% level. This revealed that the NN-I and GOR-IV methods accuracies are much influenced by the 55-65% level of Q3 prediction accuracy while the rest of the prediction methods are less influenced by this prediction level and PROF is somewhat influenced by this Q3 level.
At the 75-80% Q3 prediction level, NN-II method predicted about 180 proteins while NN-GORV-I and PROF methods predicted about 165 proteins each (Fig. 3). GOR-V predicted above 120 proteins while NN-I and GOR-IV methods predicted around 80 proteins each. This revealed that NN-II, NN-GORV-I and PROF prediction methods predicted more proteins in the 75-80% level rather than lower levels of Q3 prediction which will shift the prediction accuracies of these methods towards the high level of prediction accuracies.
At Q3 prediction level of 85-90%, NN-GORV-I and GOR-V methods predicted above 180 proteins each, while PROF predicted below 180 proteins and the NN-II method predicted around 140 proteins. GOR-IV method did not predict any number of proteins at this level and NN-I predicted around 10 proteins.
Figure 3 shows the Q3 prediction level of above 90-100% which is the highest level can be achieved to predict a protein. N NN-GORV-I method and NN-II predicted about 25 proteins each at this level. GOR-V predicted about 15 proteins while the rest three prediction methods predicted less than 10 proteins each. These results supported the suggestion that NN-GORV-I predicts many proteins at Q3 higher accuracy level compared to the other prediction methods.
In conclusion, Fig. 3 explains that the histograms distributions illustrate NN-GORV-I outperform all other classifiers or prediction methods. However, NN-I and GOR-IV are the lowest performing classifiers and GOR-V, NN-II and PROF are intermediate classifiers.
Figure 4 is a line graph designed to test the ability of these prediction methods and how they behave in the prediction of the 480 proteins. An ideal line for an ultimate predictor is a line parallel to the x-axis at a point of y-axis equal to 100. When y equals to 50 for the same parallel line then the line represents a random guess for the coils states prediction. A line travels parallel to the x-axis at y equals to 33.3 is as worst (poor) as random guess of a prediction. The results resembles the Reliability Index (RI) for predicting proteins similar to that proposed by Rost[17] that is to show the prediction methods did not only restrict their predictions to the most strongly predicted residues. It is also equivalent to the scale that discussed by Eyrich et al.[18] which plotted the accuracy versus coverage for a subset of 205 proteins.
Figure 4 shows that GOR-IV method travels from Q3 prediction accuracy near 20% and then increases steadily until it reaches 85% spinning through the 480 proteins. GOR-IV line is under all the other five lines followed by NN-I method line just above it with very minor margin following a similar pattern indicting that GOR-IV method is the poorer performing prediction method followed by NN-I method. GOR-V method, NN-II method and PROF method lines are in between the above mentioned three methods lines. GOR-V line is below the NN-II line while PROF line is above them and of course below the NN-GORV-I method line. To conclude Fig. 4 results, the newly developed method (NN-GORV-I) that combines GOR-V method and NN-II method is superior to all other methods studied in this study.
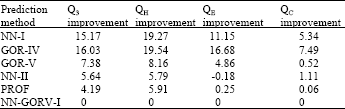
Table 1 shows the improvement of the prediction accuracy of helices, strands, coils and all the three secondary structure sates together of NN-GORV-I over the other five methods. The improvement of NN-GORV-I method over NN-I and GOR-IV is very high which is above 29% improvement for the helices and strands states but below 10% improvement for the coil states.
However, the overall performance improvement (Q3) of the NN-GORV-I method over NN-I and GOR-IV is above 15% which is a very big gain in secondary structure prediction accuracy.
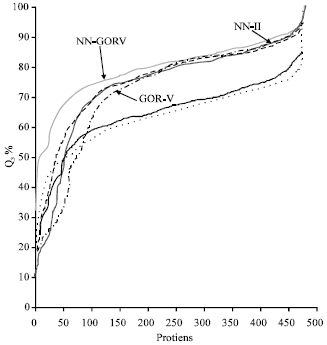 | |
| Fig. 4: | Q3 performance of the seven prediction methods |
This result is not surprising since the two low performance predictors did not implement a multiple sequence alignment method to get use of the long range interactions of residues in the amino acid sequences.
GOR-V is one of the two methods that formed the NN-GORV-I method and hence the improvement over this method is of special importance. Table 1 showed that the performance improvements of the NN-GORV-I method over GOR-V are 8.2, 5.0 and 0.5% for helices states (QH), strands (QE) and coils (QC), respectively. The improvements in helices and strands states are considerably high, especially for the strands since strands are known to be difficult to predict. The improvement in coil state is very low and this might be good sign that NN-GORV-I method is a high performance predictor since its overall gain is not from the coil states that most predictors over predict them.
When a prediction method gains an improvement in its helices and strands states, this means that this predictor is able to differentiate and discriminate between the three secondary structure states. The overall improvement (Q3) of the NN-GORV-I method over the GOR-V method is 7.4%. The reported accuracy of GOR-V is 73.5% [14] which means an improvement of about 6% is achieved. Anyhow, whatever compared to the reported accuracy of GOR-V or the calculated accuracy in this experimental study, the improvement of the NN-GORV-I method performance over GOR-V is fairly high.
NN-II method is also one of the two methods that combined NN-GORV-I method. Table 1 shows the improvements of performance of NN-GORV-I method over the NN-II method are 6.63, 8.4 and 1.66% for helices (QH), strands (QE) and coils (QC) states, respectively. The improvement of Q3 of NN-GORV-I over NN-II is 6.91%. The improvements in the helices and strand states are considerably high while the improvement in the coil states is low and as discussed before the gain in accuracies of beta strands is the most important among the three states of secondary structure. Most modern neural network methods of secondary structure prediction in the literature reported accuracies up to 70.5% and below 76.4%[19,12]. However, an overall gain of accuracy of about 5-7% in the NN-GORV-I method over NN-II is significantly high gain.
Table 1 shows that the improvements of the NN-GORV-I method over the PROF method in this experimental work are 6.75, 8.83 and 0.61% for the helices (QH), strands (QE) and c oils (QC), respectively. The 3.8-5.5% increment in the performance accuracy of the NN-GORV-I method over the PROF algorithm is considerably a significant gain in Q3 accuracy if we compare this study with the Cuff and Barton[12] where their Jnet algorithm achieved a 3.1% gain in Q3 over the PHD[12] algorithm.
Table 1 showed that the newly developed algorithm that combined the neural networks with information theory of GOR-V method is superior in performance to all method tested here in this experimental study and most method reported in the previous research.
It is important to understand that MCC is an index that shows how strong the relation between predicted and observed values. The nearest the coefficient to 1.0 the stronger the relation, while the nearest the coefficient to 0.0 the lesser the relation between observed and predicted values (Table 2).
| Table 1: | Percentage improvement of NN-GORV-I method over the other five prediction methods |
 | |
| Table 2: | Matthews correlation coefficients improvement of NN-GORV-I method over the other six prediction methods |
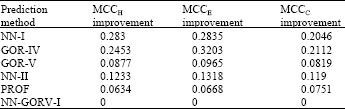 | |
There are significant improvements in the MCC o f the NN-GORV-I method over the NN-I and GOR-V methods for all the secondary structure states ranging from 0.21-0.32 which indicated that the NN-GORV-I method is significantly containing high entropy or more information to describe the relation between predicted and observed values and its prediction is of more meaning than these two methods[16,15].
As far as the improvements of the MCC of the NN-GORV-I method over the PROF method are concerned, Table 2 shows that the increments in helices, strands and coils are 0.06, 0.07 and 0.08, respectively. These are considerable improvements in the entropy of these states if we define the entropy as the information need to describe variables[16,15]. This result proved that the NN-GORV-I algorithm is not only superior in performance but also superior in describing the strength of the relations between observed and predicted states in its prediction.
CONCLUSIONS
In this study, the performance of the six methods conducted, described and assessed. The results confirmed that algorithms that did not use sequence alignment profiles like GOR-IV and NN-I are found to be of very low performance compared to other methods. When the above two methods used multiple alignment profiles and hence named GOR-V and NN-II, a significant gain in the accuracy has been achieved. The PROF method conducted in this study with almost the same database and environment of the original PROF, has achieved accuracy performance similar to that reported in the original PROF. This indicates that the statistical comparison in this study is realistic and appropriate. The NN-GORV-I algorithm outperformed the reported accuracy of the multiple cascaded classifier method (PROF).
The NN-GORV-I also proved that it is of high quality and more useful compared to the other methods. The method also proved that the entropy and the information used to describe its strength of prediction is more than the information used in the other prediction methods.
ACKNOWLEDGMENTS
The idea of this study was originated at the Artificial Intelligence (AI) lab at the Faculty of Computer Science and Information Systems (FSKSM), University of Technology Malaysia (UTM). The authors would like to thank every single person who contributed to directly or indirectly to this research.
REFERENCES
- Garnier, J., D.J. Osguthorpe and B. Robson, 1978. Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J. Mol. Biol., 120: 97-120.
CrossRefPubMedDirect Link - Garnier, J., J.F. Gibrat and B. Robson, 1996. GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol., 266: 540-553.
CrossRefPubMedDirect Link - Qian, N. and T.J. Sejnowski, 1988. Predicting the secondary structure of globular proteins using neural network models. J. Mol. Biol., 202: 865-884.
CrossRefPubMedDirect Link - Rost, B. and C. Sander, 1993. Prediction of protein secondary structure at better than 70% accuracy. J. Mol. Biol., 232: 584-599.
PubMed - Baldi, P., S. Brunak, Y. Chauvin, C.A. Andersen and H. Nielsen, 2000. Assessing the accuracy of prediction algorithms for classification: An overview. Bioinformatics, 16: 412-424.
Direct Link