
Saad Alkahtani
Department of Biology, College of Teachers, King Saud University, Riyadh, Saudi Arabia
Saud A. Alarifi
Department of Zoology, College of Science, King Saud University, Riyadh, Saudi Arabia
Amin Abdullah Al-Doaiss
Department of Zoology, College of Science, King Saud University, Riyadh, Saudi Arabia
International Journal of Zoological Research
Year: 2009 | Volume: 5 | Issue: 4 | Page No.: 161-170







ABSTRACT
The aim of the present study was to investigate the effect of antibiotic gentamicin on the apoptotic pathway in rat liver cells. For this purpose, four groups of rats were injected intramuscularly (im) with gentamicin at a dosage of 80 mg kg-1 b.wt. once daily for 1, 2, 3 and 4 weeks successively. The induced apoptosis in hepatocytes was evaluated as early as one day post-injection, the gentamicin started to induce apoptosis as assessed histologically and with the aid of Poly (ADP-ribose) polymerase (PARP) test. Histopathological examination and PARP test revealed relevant apoptosis markers at every weekly interval compared with the negative control. Irregular and condensed nuclear materials, organelles deteriorations and PARP fragmentation were the most alternations. This study demonstrated the potentiality of gentamicin to induce apoptosis in rat liver cells and the usefullness of the obtained data in daily clinical practice regarding the associated development of hepatotoxicity.
PDF Abstract XML References Citation
How to cite this article
Saad Alkahtani, Saud A. Alarifi and Amin Abdullah Al-Doaiss, 2009. Detection of Apoptotsis Induced by Gentamicin in Rat Hepatocytes. International Journal of Zoological Research, 5: 161-170.
DOI: 10.3923/ijzr.2009.161.170
URL: https://scialert.net/abstract/?doi=ijzr.2009.161.170
DOI: 10.3923/ijzr.2009.161.170
URL: https://scialert.net/abstract/?doi=ijzr.2009.161.170
INTRODUCTION
Gentamicin, an effective aminoglycoside antibiotic, is often essential for the treatment of severe infections due to Gram-negative bacteria. Since, it can affect renal functions even at any therapeutic dose, there is always a fear of an associated development of nephrotoxicity, in addition to its other side effects e.g., anemia and ototoxicity (Yasin et al., 2003). However, the treatment schedule induces nephrotoxicity, which accounts for 10B20% of acute renal failure cases (Kalayarasan et al., 2009) and leads to hearing loss (Dehne et al., 2002). Apoptosis, which is also known as programmed cell death, is an active form of cell death which plays a crucial role in the development and maintenance of cell homeostasis. Apoptosis is proved to be activated by a cascade of factors and should be placed under tight genetic regulations. It has now been recognized as an important determinant of cell degeneration in many toxic events (Yasin et al., 2003). Apoptosis may occur via a death receptor-extrinsic or intrinsic pathways. In the extrinsic pathway, Tumor Necrosis Factor (TNF) family (Fas/APO-1 ligand, TNF, TRAIL) proteins bind to the death receptors and these interactions result in conformational changes that expose a binding surface for Fas-associated Death Domain (FADD) (Lee et al., 2008). Caspases are cysteine-aspartyl-proteases that cleave a critical set of cellular proteins to initiate the apoptotic signal including several representatives involved in apoptosis with many proteins involved in the molecular signalization of apoptosis (Jung et al., 2007). Caspases are subdivided into initiator (caspase-2, -8, -9 and -10) and effector (caspase-3, -6 and -7) caspases. Their function in apoptosis is to (1) arrest the cell cycle and deactivate DNA repair; (2) to deactivate inhibitors of apoptosis and (3) to dismantle the cellular cytoskeleton. Usually, initiator caspases, once activated, will activate downstream effector caspases in a cascade-like pattern. Two main pathways are well established in apoptosis: (1) the death receptor pathway and (2) the mitochondrial pathway (Jurkiewicz et al., 2004). The Mitochondrial Permeability Transition (MPT) was first described in necrotic cell death and recent findings showed that MPT is also involved in apoptosis because cytochrome c and the apoptosis-inducing factor normally located at the intermembrane space of mitochondria are released into the cytosol where they are able for example to activate the caspase cascade (Dehne et al., 2002). Earlier studies have shown that mitochondria play a central role in cell death in response to DNA damage (Lee et al., 2008).
Among the various subcellular organelles potentially involved in apoptosis, both lysosomes and mitochondria have been shown to send death signals through the activation of specific stress sensors. For mitochondria, two main events leading to apoptosis which are the disruption of the transmembrane potential and the release of cytochrome c, which contribute to formation of the so-called apoptosome leading to the successive activation of caspase-9 and of the executioner caspase-3 (Servais et al., 2005). The list of intracellular caspase substrates is still growing and caspase-3 is known to cleave a large number of them such as PARP which is a nuclear enzyme that is responsible for catalyzing the transfer of ADP-ribose polymers onto itself and other nuclear proteins in response to DNA strand breaks. Also, PARP cleavage is considered as a signature event for apoptosis (Jurkiewicz et al., 2004).
The majority of such cell deaths share common characteristics, such as cell shrinkage, bubbling of the plasma membrane, chromatin condensation, apoptotic bodies and finally fragmentation of DNA as a biochemical hallmarks of apoptosis (Ho et al., 1999).
The present investigation was undertaken in an effort to identify the apoptotic process which could be induced by the antibiotic gentamicin in the hepatocytes of rat. The apoptotic changes were described at the ultrastructure level and PARP cleavage was employed as a biochemical mark.
MATERIALS AND METHODS
All of the experimental procedures were conducted in the Molecular Biology Lab., of the King Saud University, Saudi Arabia between 2008 and 2009.
Experimental Animals
Apparently healthy male Wistar albino rats, 8-10 weeks old and weighing 220-250 g, were obtained from the animal house facilities of the Science College, King Saud University in Riyadh and kept in stainless-steel cages, 5 rats per cage and maintained under standard laboratory conditions at a temperature of 22±11C, a relative humidity of 45±5% and photoperiod cycle of 12/12 h. Rats were fed a commercial pellet diet and water was offered ad libitum.
Experimental Design
A total of 25 males were used and divided randomly into five groups designated A, B, C, D and E, each group contained 5 males. Group-A were treated orally with distilled water (2 mL kg-1) and injected intramuscularly (im) for with 0.9% NaCl (sterile normal saline) at a dose of 2 mL kg-1 a day as a negative control. Groups-B, C, D and E were injected as in group-A except that the normal saline was replaced with gentamicin at a dose of 80 mg kg-1 b.wt. a day for various periods: one week (group-B), two weeks (group-C), three weeks (group-D) and four weeks (group-E).
Gentamicin
Gentamicin was obtained from the Saudi Drugs and Medical Instruments Company (SPIMA Co.), Buraydah, Saudi Arabia.
Antibodies
Primary Antibody Anti-Poly (ADP-Ribose) Polymerase (PARP)
The primary antibody was obtained from Cell Signalling Co., USA. and used to detect the intact PARP (116 kDa) enzyme, as well as the large (89 kDa) and small (24 kDa) fragments produced following hydrolysis of intact PARP with caspase-3. The polyclonal antibody was produced by immunizing rabbits and diluted (1:1000) with 5% skimmed milk in PBS containing 0.1% Tween 20.
Secondary Antibody (Anti-Rabbit IgG, HRP-Linked Antibody)
The secondary antibody was obtained from Cell Signalling Co., USA. The secondary antibody was labeled with peroxidase and assayed using the enhanced chemiluminescence (ECL) method. Western blotting detection reagents were obtained from Amersham Co., RPN2106PC, USA.
Protein Extraction
Approximately 10 g from each experimental rat liver was homogenized in a cold homogenizer tube containing 2 mL of homogenization buffer. Homogenates were spun at 300 rpm for 10 min and the supernatants were removed and stored at -80°C. The concentration of total protein in each sample was estimated spectrophotometrically (GeneQuant pro, Amersham Co., USA) at a wave length measuring 595 nm. Equal volumes of 2X sample buffer and protein (30 Fg FL-1) were mixed in an Eppendorf tube and heated to 95°C for 5 min before loading. The process protein extraction was done according to the method described by Hossain et al. (2000) and Mathas et al. (2003).
SDS-PAGE and Immunoblotting
The mixture of proteins and 2X sample buffer were separated on 30% polyacrylamide gel using a PowerPac Basic system (S.N 37S/7159, Italy) at 50 V for 1 h and then at 100 V by the end of electrophoresis. Proteins were then blotted on nitrocellulose membrane. The nitrocellulose membrane was washed several times with Phosphate Buffered Saline (PBS) and then incubated in 5% skimmed milk in PBS containing 0.1% Tween 20 to block non specific sites. Membranes were incubated with the primary antibody (Anti-PARP) overnight at 4°C and then with the secondary antibody for 3 h. Membranes were then washed three times (5 min each) in PBS-T. All steps were done under a mild agitation. Protein bands were visualized using SuperSignal West Pico Chemiluminescence Substrate according to the manufacturers instructions. The molecular size of the visualized protein bands was determined by comparison with standard markers (Moronvalle-Halley et al., 2005; Servais et al., 2005).
Electron Microscopy
Liver tissues specimens collected from all experimental animals were immediately fixed by immersion in 3% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4 for 12 h at 4°C. Tissues specimens were post-fixed in 1% osmium tetroxide (OsO4) and then dehydrated in ascending grads of ethanol and finally embedded in Epon resin mixture. Ultrathin sections are (70-80 nm) were prepared using Leica UCT ultramicrotome and contrasted with uranyl acetate and lead citrate. The contrasted ultrathin sections were examined and photographed under a transmission electron microscope (TEM, JEOL 1010, Japan) (El-Mouedden et al., 2000).
RESULTS
Ultrastructure Findings
Hepatic cells of control rats revealed the normal ultrastructure appearance including the regular nucli and intact cytoplasmic organelles (Fig. 1). Hepatic cells from rats injected with gentamicin at all week intervals revealed the ultrastructural alternations typical of apoptosis. The cells were shrunk displaying irregular nuclei having segregated chromatin. Apoptotic bodies containing intact organelles were also detected and some cells with swollen mitochondria were noted (Fig. 2, 3). In addition to these alternations related to apoptosis, some necrotic cells were observed (Fig. 4). None of these morphological abnormalities were noted in control cells. The frequency of apoptotic changes increased in rats injected for long duration.
Poly (ADP-Ribose) Polymerase (PARP)
The PARP was evaluated by western blotting analysis as shown in Fig. 5, all treatments with various doses of gentamicin induced apoptosis and yielded positive results (B, C and D) in terms of the degradation of intact PARP molecules (116 kDa) to generate the large (89 kDa) fragments (Fig. 5) also shows that treatments produced different bands which increased gradually in density with increasing treatment durations, while no degradation was observed in negative control panel (A). Cleavage of PARP into a 89 kDa fragment appeared as early as the first week of gentamicin treatment. This result of PARP degradation proved that gentamicin can induce apoptosis in rat liver cells.
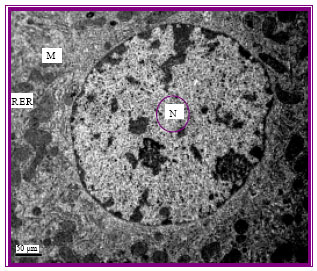 | |
| Fig. 1: | The normal control rat hepatic cell. Note the normal regularly round nucleus (N) containing clearly visible nucleolus, the dispersed euchromatin and clumped heterochromatin beneath the inner nuclear membrane. Also, mitochondria (M) and Rough Endoplasmic Reticulum (RER) are morphologically normal x6000 |
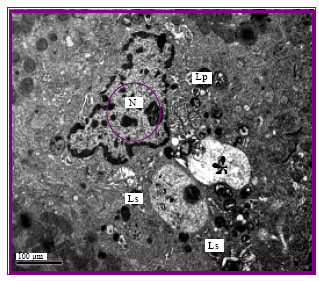 | |
| Fig. 2: | Hepatic cell of gentamicin-injected rat showing the typical apoptotic alteration Nucleus (N) with irregular condensed nuclear material and other organelles are deteriorated (*). There is a large number of lysosomes (Ls) and lipid droplets (Lp) x5000 |
 | |
| Fig. 3: | Hepatic cell of gentamicin-injected rat showing mitochondrial alteration including swelling, disruption of mitochondrial cristae. The lateral mitochondria appear as vacuolar structures x8000 |
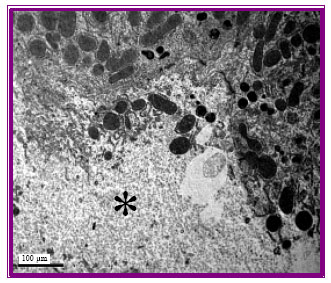 | |
| Fig. 4: | Liver tissue of gentamicin-injected rat showing necrotic focus (*) with complete alteration of organelles x8000 |
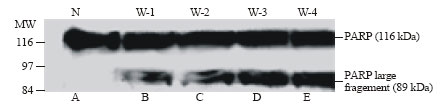 | |
| Fig. 5: | Western blot analysis of PARP from livers of rats injected with gentamicin (80 mg kg-1) (groups B-E). N: Control (A). Marker (MW) |
DISCUSSION
The obtained data showed that gentamicin induced apoptosis in rat liver cells as evidenced by morphological changes of liver cells and PARP test compared with negative control. This elucidates its potential to induce cytotoxicity. Furthermore gentamicin-induced nephrotoxicity has been well established in a rat model, which accounts for 10B20% of the cases of acute renal failure in rats. In gentamicin-induced nephrotoxicity, Reactive Oxygen Species (ROS) is believed to play a pivotal role in cellular damage and necrosis via several complex mechanisms including peroxidation of membrane lipids, protein denaturation and DNA damage. It was reported that gentamicin acts as an iron chelator and that the ironBgentamicin complex is a potent catalyst of free radical formation (Kalayarasan et al., 2009). Gentamicin seems to generate (ROS) in cell-free as well as in cellular systems and induce the opening of the mitochondrial permeability pore and, in addition, might exert direct effects at the plasma membrane, potentially activating the Jun kinase pathway (Dehne et al., 2002).
Generally, ROS and cytotoxins can cause cell death by necrosis or apoptosis, often in a dose-dependent manner. High dosages of the toxicant usually result in necrosis characterized by progressive cell and organelle membrane dysfunction, leading to loss of ion homeostasis and secondly to the inability to maintain mitochondrial respiration and ATP levels essential for cellular survival. On the other hand, moderate doses of cytotoxins or ROS can activate the apoptotic pathway (Dehne et al., 2002). Oxidative stress is the inappropriate exposure to ROS and results from the imbalance between prooxidants and antioxidants leading to cell damage and tissue injury. The ROS generation is increased in many pathological situations. In liver diseases, an excess of ROS can induce cell death by either apoptosis or necrosis. Apoptosis, or programmed cell death is an active process characterized by cell shrinkage, chromatin condensation, formation of apoptotic bodies and activation of caspases. In contrast, necrosis is passive and associated with ATP depletion, rupture of the plasma membrane and spilling of the cellular content eliciting inflammation (Conde de la Rosa et al., 2006). Gentamicin accumulates in lysosomes and induces apoptosis in kidney proximal tubules and renal cell lines (Servais et al., 2005). In studying aminoglycoside toxicity in vivo, morphological alterations associated with apoptosis have been found in cochlear hair cells and in vestibular hair cells using electron microscopy and in situ nick end labeling showed DNA fragmentation and that was confirmed by examining the morphological alterations and the membrane potential of mitochondria during gentamicin-induced cell damage using the in vitro model of the cochlear neurosensory epithelium. There are indications for an involvement of mitochondria in ototoxicity because inhibition of mitochondrial protein synthesis increases gentamicin toxicity in vivo and some mutations of mitochondrial DNA increase the sensitivity to aminoglycosides in humans (Dehne et al., 2002). The high concentration of aminoglycoside caused cell damage within 2 h. Therefore the concentration used (1 mM) to cause significant cell death is slightly higher than the concentrations used in the studies with mouse or rat organ cultures (0.1 mM netilmicin or neomycin). In addition, isolated OHCs from the guinea pig cochlea are not damaged by native gentamicin, not even by incubation with gentamicin concentrations as high as 10 mM. In order to damage isolated cells, gentamicin first has to be >activated= to a metabolite. Since, our data show that gentamicin induced apoptosis in cochlear OHCs under the conditions used, our model seems to be suitable to study the pathomechanisms of gentamicin toxicity in vitro (Dehne et al., 2002).
We do know whether the triggering of the mitochondrial pathway is due to a direct effect of the antibiotic on mitochondria, or whether it results from some other mechanism. The first hypothesis implies that gentamicin has access to the mitochondria, which may simply result from its release from lysosomes as suggested in previous studies. It is interesting to note, that gentamicin is capable of permeabilizing liposomes with a composition that mimics that of the outer mitochondrial membrane at pH 7.4. While this process is less rapid than at pH 5.4, it is nevertheless faster than what is observed for liposomes of a composition mimicking the inner leaflet of the mitochondrial membrane. The latter observation shows that aminoglycosides have a larger affinity for phosphatidylinositol (one of the acidic phospholipids present in lysosomal and outer mitochondrial membrane) than for cardiolipin (which is abundant in the inner mitochondrial membrane (Servais et al., 2005). The cellular changes induced by apoptosis occur after a cascade of cell signalling and caspase-mediated events regulated by both pro- and anti-apoptotic proteins and are triggered by two major pathways: the death-receptor-induced pathway or extrinsic pathway and the mitochondria-apoptosome-mediated pathway or intrinsic pathway. The extrinsic pathway implicates death ligands such as Fas ligand, TNFα, TRAIL and their receptors. The intrinsic pathway includes apoptotic stimuli induced by cytotoxic drugs or oxidative stress which targeted mitochondria. This pathway involves the release of cytochrome c from the mitochondria to the cytosol, which induces apoptosome complex formation and results in protease procaspase-9 activation and subsequent activation of procaspase-3 through proteolytic cleavage visualized by the decrease of proform level and appearance of cleavage products. Both the extrinsic and intrinsic apoptotic pathways lead to caspase-3 activation and cleavage of a limited set of essential cellular proteins, leading to cell dismantlement. In the liver, the apoptosis induced by TAA could result from a combination of both pathways: the intrinsic apoptosis pathway by generation of oxidative stress and the extrinsic apoptosis pathway by activation of Kupffer cells which can secrete TNF (Moronvalle-Halley et al., 2005). Several intranucleolus changes produced from activation of caspases enzymes such as active DNase, PARP and Lamina-A degradation as apoptosis markers (Kang et al., 2006; Mclaren et al., 2006; Yu et al., 2008) and many studies published on PARP sensitivity and its response to apoptosis (Qin et al., 2008; Ochi et al., 2008). The activation and cleavage of PARP was investigated in various human cancer cells treated with chemotherapeutic drugs (Ho et al., 1999) and as a result of PARP activation which resulted from early DNA damage response, NAD+ levels may rapidly decline, which may affect the activity of the enzymes involved in glycolysis and the Krebs cycle. In an attempt to restore NAD+ pools cell resynthesized NAD+ by combining nicotinamide with 2ATP and as a consequence cellular ATP levels become depleted and a cellular energy crisis may arise leading to cell death. Cells that are replicating and growing and utilizing almost exclusively glucose die from NAD+ and ATP depletion as a consequence of PARP activation (Brock et al., 2004; Mikami et al., 2004; Shi et al., 2004; Wijk and Hageman, 2005). When DNA is moderately damaged, PARP participates in the DNA repair process and cells survive. However, in the case of extensive DNA damage, PARP overactivation induces a decrease of NAD+ and ATP levels, leading to cell dysfunction or even to necrotic cell death and pathogenesis of several diseases. Despite the important role of PARP to detect apoptosis, some evidence suggests the involvement of PARP in necrosis and during apoptosis PARP activity is suppressed and the cells are forced to die because of the mild effect of toxis and this mechanism needs enough energy to support apoptosis. But the necrosis is more severe than apoptosis and caused by severe genotoxic stimuli and this kind of cell death takes place in a tissue or organ. There are several biochemical and morphological differences between apoptosis and necrosis (Ho et al., 1999; Nguewa et al., 2003). Cells exposed to DNA-damaging agents may undergo three pathways depending on the degree of DNA damage. Thus, mild DNA damage activates PARP, which subsequently interacts with several proteins involved in DNA repair such as polymerase II and DNA ligase III. If DNA repair proceeds successfully, then the cell survives. If DNA damage is too severe to be repaired, apoptosis take place, so that the caspase cleaves PARP. A third pathway may be induced by extensive DNA breakage in which overactivation of PARP cleaves NAD+ into NAD and ADP-ribose moieties and polymerizes the later onto nuclear acceptor proteins and a decrease of NAD+ levels inhibits production of ATP through oxidative phosphorylation, leading to ATP depletion and necrotic cell death. As mentioned, within a population of tumor cells, necrosis and apoptosis may take place together in response to cytotoxic drugs. This may be attributable to drug concentration reaching different cancer cells; low concentration induce apoptosis and higher concentration produce necrosis (Nguewa et al., 2003). Induced cell death plays a fundamental role in toxicant-induced liver damage and this has been described as necrotic or apoptotic although more recently these distinctions are blurring. In addition, It has been recently hypothesized that necrosis and apoptosis are not separate processes; rather they are the opposite extremes of a common pathway (Moronvalle-Halley et al., 2005).
CONCLUSION
The present study demonstrates the potential of gentamicin to induce apoptosis in rat liver cells. On the other hand the liver has the ability to regenerate and rapidly clear eliminate apoptotic cells in vivo and the apoptosis was induced more rapidly in the liver than in other tissues observed in vivo. According to applied dose gentamicin also is useless in daily clinical practice because its associated with toxicity development, in addition to its other side effect. This may be one of the manifestations of the toxicity of gentamicin, but because this antibiotic is often essential for the treatment of infections due to Gram-negative bacteria further, studies are needed to clarify the suitable daily dosage and its effects on human tissues in vivo.
ACKNOWLEDGMENTS
The authors express their deep gratitude to Dr. Mohammed Mubarak (Electron Microscope Unit, College of Medicine, King Saud University, Riyadh, Saudi Arabia) for his valuable assistance in identifying the markers of apoptosis and Dr. Ahmed Ali Al-Qahtani (Department of Biological and Medical Research, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia) for his invaluable assistance during the immunobloting study.
REFERENCES
- Brock, W., L. Milas, S. Bergh, R. Lo, C. Szabo and A. Mason, 2004. Radiosensitization of human and rodent cell lines by INO-101, a novel inhibitor of poly (ADP-ribose) polymerase. Cancer Lett., 205: 155-160.
CrossRef - Dehne, N., U. Rauen, H. Groot and J. Lautermann, 2002. Involvement of the mitochondrial permeability transition in gentamicin ototoxicity. Hear. Res., 169: 47-55.
CrossRef - El-Mouedden, M., G. Laurent, M.P. Mingeot-Leclercq and P.M. Tulkens, 2000. Gentamicin-induced apoptosis in renal cell lines and embryonic rat fibroblasts. Toxicol. Sci., 56: 229-239.
PubMed - Ho, Y., H. Lee, C. Chang and J. Lin, 1999. Induction of bax protein and degradation of lamin A during p53-dependent apoptosis induced by chemotherapeutic agents in human cancer cell lines. Biochemical Pharmacol., 57: 143-154.
CrossRef - Hossain, K., A. Akhand, M. Kato, J. Due and K. Takeda et al., 2000. Arsenite induces apoptosis of murine T lymphocytes through membrane raft-linked signaling for activation of c-Jun amino-terminal kinase. J. Immunol., 165: 4290-4297.
PubMed - Jung, J.Y., C.R. Han, Y.J. Jeong, H.J. Kim and H.S. Lim et al., 2007. Epigallocatechin gallate inhibits nitric oxide-induced apoptosis in rat PC12 cells. Neurosci. Lett., 411: 222-227.
CrossRefDirect Link - Jurkiewicz, M., D. Averill-Bates, M. Marion and F. Denizeau, 2004. Involvement of mitochondrial and death receptor pathways in tributyltin-induced apoptosis in rat hepatocytes. Biochimica Biophysica Acta (BBA)-Molecular Cell Res., 1693: 15-27.
CrossRef - Kalayarasan, S., P. Nagendraprabhu, N. Sriram, R. Manikandan, M. Arumugam and G. Sudhandiran, 2009. Diallyl sulfide enhances antioxidants and inhibits inflammation through the activation of Nrf2 against gentamicin-induced nephrotoxicity in Wistar rats. Eur. J. Pharmacol., 606: 162-171.
CrossRef - Kang, H.M., S.K. Lee, D.S. Shin, M.Y. Lee and D.C. Han et al., 2006. Dehydrotrametenolic acid selectively inhibits the growth of H-ras transformed rat2 cells and induces apoptosis through caspase-3 pathway. Life Sci., 78: 607-613.
CrossRef - Lee, D.H., M. Szczepanski and Y. Lee, 2008. Role of Bax in quercetin-induced apoptosis in human prostate cancer cells. Biochemical Pharmacol., 75: 2345-2355.
CrossRef - Mathas, S., A. Lietz, M. Janz, M. Hinz and F. Jundt et al., 2003. Inhibition of NF-kB essentially contributes to arsenic-induced apoptosis. Am. Soc. Hematol., 102: 1028-1034.
PubMed - Mclaren, S.H., D. Gao, L. Chen, R. Lin and J.R. Eshleman et al., 2006. Oxidative stress and DNA damage-DNA repair system in vascular smooth muscle cells in artery and vein grafts. J. Cardiothroracic Renal Res., 1: 59-72.
CrossRef - Mikami, O., S. Yamamoto, N. Yamanaka and Y. Nakajima, 2004. Porcine hepatocyte apoptosis and reduction of albumin secretion induced by deoxynivalenol. Toxicology, 204: 241-249.
CrossRef - Moronvalle-Halley, V., B. Sacre-Salem, V. Sallez, G. Labbe and J.C. Gautier, 2005. Evaluation of cultured, precision-cut rat liver slices as a model to study drug-induced liver apoptosis. Toxicology, 207: 203-214.
CrossRef - Nguewa, P., M. Fuertes, C. Alonso and J. Perez, 2003. Pharmacological modulation of Poly(ADP-ribose) polymerase-mediated cell death: Exploitation in cancer chemotherapy. Mol. Pharmacol., 64: 1007-1014.
PubMed - Ochi, T., K. Kita, T. Suzuki, A. Rumpler, W. Goessler and K. Francesconi, 2008. Cytotoxic, Genotoxic and cell-cycle disruptive effects of thio-dimethylarseinate in cultured human cells and the role of glutathione. Toxicol. Applied Pharmacol., 228: 59-67.
CrossRef - Qin, X., L. Hudson, W. Liu, G. Timmins and K. Liu, 2008. Low concentration of arsenite exacerbates UVR-induced DNA strand breaks by inhibiting PARP-1 activity. Toxicol. Applied Pharmacol., 232: 41-50.
CrossRef - Conde de la Rosa, L., M. Schoemaker, T. Vrenken, M. Buist-Homan, R. Havinga, P. Jansen and H. Moshage, 2006. Superoxide anions and hydrogen peroxide induce hepatocyte death by different mechanisms: Involvement of JNK and ERK MAP kinases. J. Hepatol., 44: 918-929.
CrossRef - Servais, H., P. Smissen, G. Thirion, G. Essen, F. Bambeke, P. Tulkens and M. Mingeot-Leclercq, 2005. Gentamicin-induced apoptosis in LLC-PK1 cells: Involvement of lysosomes and mitochondria. Toxicol. Applied Pharmacol., 206: 321-333.
CrossRef - Shi, H., L. Hudson and K. Liu, 2004. Oxidative stress and apoptosis in metal ion-induced carcinogenesis. Free Radic. Biol. Med., 37: 582-593.
CrossRef - Wijk, S. and G. Hageman, 2005. Poly(ADP-ribose)polymerase-1 mediated caspase-independent cell death after ischemia/reperfusion. Free Radic. Biol. Med., 39: 81-90.
CrossRef - Yasin, K.F., M. Sabir, M.I.K. Sherwani, M. Zafar, S. Yasmin and M.I. Alam, 2003. Amelioration of Gentamicin Nephrotoxicity by vitamin B6 (a general and histochemical profile). Pak. J. Med. Res., 42: 69-73.
Direct Link - Yu, Q.T., M. Saruta and K.A. Papadakis, 2008. Visilizumab induces apoptosis of mucosal T lymphocytes in ulcerative colitis through activation of caspase 3 and 8 dependent pathways. Clin. Immunol., 127: 322-329.
CrossRef