
Yogesh Chander
Department of Veterinary Population Medicine, University of Minnesota, Saint Paul, MN 55108, USA
Ashok K. Tiwari
Department of Veterinary Population Medicine, University of Minnesota, Saint Paul, MN 55108, USA
Suchitra Sajja
Department of Veterinary Population Medicine, University of Minnesota, Saint Paul, MN 55108, USA
M. A. Ramakrishnan
Department of Veterinary Population Medicine, University of Minnesota, Saint Paul, MN 55108, USA
Kay S. Faaberg
Department of Veterinary and Biomedical Sciences, College of VeterinaiY Medicine, University of Minnesota, Saint Paul, MN 55108, USA
Sagar M. Goyal
Department of Veterinary Population Medicine, University of Minnesota, Saint Paul, MN 55108, USA
International Journal of Virology
Year: 2007 | Volume: 3 | Issue: 3 | Page No.: 100-106







ABSTRACT
Feline calicivirus (FCV) is one of the most common pathogens of cats causing upper respiratory tract infections. A real time RT-PCR assay was developed for the rapid detection of FCV. A set of probe/primers was designed to amplify a region of 151 bp based on the conserved region of 7 FCV strains (FCV 255, 2280, NADC, F9, KCD, CF I and Urbana) by sequence comparison. The assay was found to be more sensitive than virus isolation and was linear over a wide range of template concentrations.
PDF Abstract XML References
How to cite this article
Yogesh Chander, Ashok K. Tiwari, Suchitra Sajja, M. A. Ramakrishnan, Kay S. Faaberg and Sagar M. Goyal, 2007. A TaqManR RT-PCR Assay for the Detection of Feline calicivirus. International Journal of Virology, 3: 100-106.
DOI: 10.3923/ijv.2007.100.106
URL: https://scialert.net/abstract/?doi=ijv.2007.100.106
DOI: 10.3923/ijv.2007.100.106
URL: https://scialert.net/abstract/?doi=ijv.2007.100.106
INTRODUCTION
Feline calicivirus (FCV) is a single stranded RNA virus belonging to the family Caliciviridae and is a common cause of upper respiratory tract infection in cats (Gaskell and Dawson, 1994). In addition, FCV may also cause conjunctivitis, stomatitis, enteritis and lameness (Baulch-Brown et al., 1997). Infections caused by strains of low virulence are usually restricted to oral mucosa, nasal passages and conjunctiva while those by virulent and pneumonic strains show signs of severe respiratory infection. Mortality is high in the presence of secondary bacterial infections (Murphy et al., 1999). Recovered cats can serve as carriers of the virus and are important in the maintenance and dissemination of infection in domestic and wild cat populations. Harbour et al. (1991) reported virus isolation rates of 20% from oropharyngeal swabs collected from domestic cats without any breed or sex variation. A similar rate of high FCV carriage was reported from a cat shelter and purebred cattery in California (Tenorio et al., 1991). Although the duration of shedding is variable, approximately 50% of the infected cats cease virus shedding by 75 days post-infection although individual cats may shed virus for up to 2 years (Barlough, 1992).
Clinical diagnosis of FCV is based on history and clinical signs but definitive diagnosis requires virus isolation in cell cultures and/or serology. Since virus isolation procedures are time consuming and expensive, tests based on nucleic acid amplification (e.g., reverse transcription-polymerase chain reaction or RT-PCR) have been developed. However, these tests have been hampered by difficulty in designing oligonucleotide primers that are able to amplify nucleic acids from all different strains of FCV (Sykes et al., 1998). In addition, PCR products need to be detected by gel electrophoresis or Southern blotting thus increasing the time and expense in the specific detection process. Recently, Marsilio et al. (2005) reported on a nested PCR (nPCR) for the detection of FCV. Although more specific than RT-PCR, nPCR requires the use of two sets of primers and two cycles of PCR followed by electrophoretic separation of bands, which again increases the time as well as cost of diagnosis.
Recently, real time-PCR has become a method of choice for pathogen detection because it allows real time quantification of PCR products, thus eliminating the need for any post-amplification steps (Leutenegger, 2001). Fluorogenic PCR assays have been successfully used in amplification and detection of various bacterial and viral pathogens (Batt, 1997). Gut et al. (1999) described a one-tube fluorogenic, RT-PCR for the quantification of feline corona viruses. Schweiger et al. (2000) reported the development of a TaqManR RT-PCR for typing and subtyping of influenza viruses in the human clinical samples. This test was more sensitive than viral isolation as it could detect up to 0.1 TCID50 of the virus. The TaqMan assay employs 5 → 3exonuclease activity of Thermus aquaticus (Taq) DNA polymerase to hydrolyze an internal TaqMan probe labeled with a florescent reporter dye (FAM) and a quencher dye (TAMRA). The most striking advantage of TaqMan assay over conventional RT-PCR is the ability to save time as it allows real time quantification of amplification products. In this report, we describe the development of a TaqManR assay for the detection of FCV in clinical samples.
MATERIALS AND METHODS
Viruses and Cells
This study was done at the Veterinary Diagnostic Laboratory, University of Minnesota, Saint Paul, MN. All seven FCV strains (FCV 255, 2280, NADC, F9, KCD, CF I and Urbana) used in this study were obtained from Dr. John Neill, National Animal Disease Center, Ames, Iowa. Monolayers of Crandell-Reese feline kidney cells (CRFK) were used to propagate and titrate FCVs. Briefly, the cells were grown in Eagle’s minimal essential medium (MEM; Celox, St. Paul, MN) supplemented with 8% fetal bovine serum, penicillin (100 U mL-1), streptomycin (100 μg mL-1), fungizone (1 μg mL-1), 15 mM HEPES and 5 mg mL-1 lactalbumin hydrolysate. Confluent monolayers of CRFK cells were inoculated with the virus and after adsorption for 1 h, the cells were incubated in the maintenance medium (MEM without fetal bovine serum) at 37°C until the appearance of cytopathic effects (CPE), usually within 2-3 days post-infection. The viruses were harvested when CPE was observed in 60-80% cells. For virus harvesting, the cultures were frozen and thawed twice followed by centrifugation at 2500 x g for 10 min at 4°C. Clarified supernatant was aliquoted in 1 mL amounts and stored at -70°C until further use. For virus titration, serial 10 fold dilutions of viruses were prepared in maintenance medium and inoculated in 96 well plates containing monolayers of CRFK cells. Virus titers were calculated by the method of Reed and Muench (1938).
RNA Extraction
Viral RNA was prepared from 140 μL of cell-free extract using the Viral RNA extraction kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. RNA was eluted from silica columns in 60 μL of nuclease-free water and stored at -70°C until further use.
Primer and Probe Design
Oligonucleotide primers were derived from conserved regions identified by aligning the sequences of capsid protein gene of 7 FCV strains. Details of strains used and the conserved regions are given in Table 1. Nucleotide sequences were retrieved from GenBank (http://www.ncbi.nlm.nih.gov) and aligned using ClustalX method to determine the conserved region. Primers and probe were designed using Primer Express software and were specific to FCV 255 (Table 2). Specificity of both primers and probe for other FCV strains was determined by BLAST analysis.
Real Time RT-PCR
Real time RT-PCR (rRT-PCR) was done using the TaqMan one step RT-PCR Master Mix kit (Applied Biosystems, CA). Each reaction mixture consisted of 12.5 μL of 2X Master Mix; 0.625 μL of 40X Multiscribe Mix; 0.4 μM of each primer; 0.8 μM of TaqMan probe; 5 μL of template RNA; and 2.875 μL of water to make a final volume of 25 μL rRT-PCR was performed in 96 well plates (Applied Biosystems, CA) using the ABI PRISM 7900 sequence detection system.
| Table 1: | Sequence alignment of different Feline calicivirus (FCV) strains showing homologous region |
 | |
| Table 2: | Nucleotide sequence of probe and primers used in real time RT-PCR |
 | |
Thermocycling conditions used were: reverse transcription (RT step) at 48°C for 30 min; AmpliTaq Gold activation at 95°C for 10 min and 50 cycles of PCR consisting of denaturation and annealing at 95°C for 15 sec and 50°C for 1 min, respectively.
Specificity and Sensitivity of Assay
The sensitivity of the TaqMan assay was determined by testing RNA extracts from serial 10 fold dilutions of FCV 255 (initial titer 1.45x109 TCID50 mL-1) as a template. For standard curve, serial 10 fold dilutions of RNA extracted from undiluted virus were made in DNase and RNase-free water and then used as a template in the TaqMan assay. The detection threshold was based on the lowest concentration of viral RNA which could be detected and remained within the linearity of the standard curve. Specificity of the primers and probe was evaluated using CHV (Canine herpes virus) and FPV (Feline panleukopenia virus) isolates.
Field Isolates
Samples of trachea, spleen, lung and regional lymph nodes are routinely submitted to the Minnesota Veterinary Diagnostic Laboratory for the detection of FCV. For isolation of FCV, tissue homogenates from these samples are inoculated into CRFK cells and observed for development of CPE for up to 7 days. Samples positive for CPE are examined by negative contrast electron microscopy (Goyal et al., 1987). A total of 20 such samples were tested with rRT-PCR and the results were compared with conventional virus isolation method.
RESULTS
Analytical Specificity of rRT-PCR
The designed probe/primer set was able to amplify target viral RNAs from all 7 FCV strains used in the present study. Maximum CT (threshold cycle) value was obtained for F9 strain of FCV while the minimum value was for NADC strain. The PCR products were separated on 1.5% agarose gel and a single band at 151 bp position was observed for all seven strains (Fig. 1). The primer/probe set failed to amplify this region in CHV and FPV.
Field Samples
When 20 field samples were tested, 8 were found to be positive for FCV by the conventional virus isolation method. Of these, 7 were found to be positive by rRT-PCR.
Amplification Efficiency of rRT-PCR
Amplification efficiency of the assay was determined by using serial 10 fold dilutions of viral RNA from FCV 255 (85 μg mL-1) as the PCR template. The assay was found to detect viral RNA up to 10-7 dilution (8.5 pg mL-1) over the range of linearity with slope value of -3.29 and R2 (coefficient of correlation) 0.993 (Fig. 2). When RNA extracts of serial 10 fold dilutions of FCV 255 (initial titer 1.45x109 mL-1) were used as PCR template, the assay was able to detect RNA from virus suspension diluted to the equivalent of 0.01 TCID50.
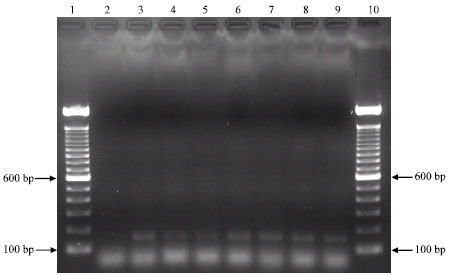 | |
| Fig. 1: | Agarose gel separation of amplification products of different FCV strains after real time RT-PCR. Lane 1 and 10: Molecular marker; Lane 2: Feline panleukopenia virus; Lane 3: FCV 255; Lane 4: F9; Lane 5: Urbana; Lane 6: FCV 2280; Lane 7: NADC; Lane 8: FCV KCD; Lane 9: FCV CFI |
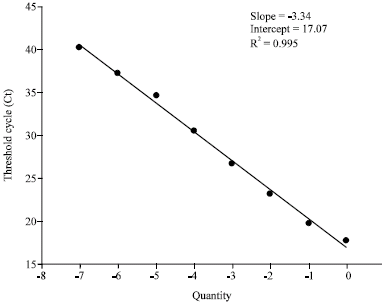 | |
| Fig. 2: | Standard curve obtained with serial 10 fold dilutions of FCV 255 RNA. Ct values are plotted against different dilutions of viral RNA, when used as template for real time RT-PCR |
DISCUSSION
As with many other RNA viruses, FCV exhibits considerable antigenic heterogeneity during replication in its host. Amino acid substitutions have been shown to occur between residues 426 and 458 of FCV capsid protein E region indicating that serotypic determinants of FCV important for antigenic variation are contained in this region (Kreutz et al., 1998). Thus it is important to develop a reliable, inexpensive and rapid system for the detection of all different FCV strains.
We describe the development of an rRT-PCR assay for rapid detection of FCV with high sensitivity and specificity. The assay was able to detect as little as 0.01 TCID50 of FCV. The sensitivity of nPCR protocol described by Marsilio et al. (2005) was 1000 fold greater than virus isolation in cell culture (nPCR was positive at 10-11 dilution of stock virus whereas virus isolation was positive to 10-8 dilution). However, nPCR requires the use of two sets of primers for specific amplification followed by separation of PCR products on agarose gel thus increasing time of detection and risk of contamination. In our protocol, the rRT-PCR is completed within 3 h without any need for the analysis of PCR products, thus reducing the time of diagnosis. Helps et al. (2002) reported an rRT-PCR for detection of FCV by melting curve analysis using SYBR green I dye which was more sensitive than conventional PCR as this could detect wide range of field isolates. However, this method is not as sensitive as TaqMan assay since SYBR green dye binds to any double stranded DNA produced by reverse transcription resulting in non-specific products (Helps et al., 2002). The set of probe/primers used in this study did not detect CHV and FPV suggesting that the described rRT-PCR protocol is specific for the detection of FCV.
The newly developed rRT-PCR was able to detect 7 of 8 field samples this assay may not detect all FCV isolates. Helps et al. (2002) noted that FCV isolates are highly variable and that no single probe is able to detect all the isolates. Although a major step forward in the rapid detection of FCV, we believe that studies need to be continued to develop single unified tests that can detect all field strains of FCV. Since the detection limit of rRT-PCR is 0.01 TCID50, this test should be useful in detecting carrier animals that are known to shed small quantities of virus.
REFERENCES
- Gut, M., C.M. Leutenegger, J.B. Huder, N.C. Pedersen and H. Lutz, 1999. One tube fluorogenic reverse transcription-polymerase chain reaction for the quantification of Feline coronaviruses. J. Virol. Methods, 77: 37-46.
CrossRefDirect Link - Helps, C., P. Lait, S. Tasker and D. Harbour, 2002. Melting curve analysis of Feline calicivirus isolates detected by real-time reverse transcription PCR. J. Virol. Methods, 106: 241-244.
CrossRefDirect Link - Kreutz, L.C., R.P. Johnson and B.S. Seal, 1998. Phenotypic and genotypic variation of Feline calicivirus during persistent infection of cats. Vet. Microbiol., 59: 229-236.
Direct Link - Marsilio, F., B.D. Martino, N. Decaro and C. Buonavoglia, 2005. A novel nested PCR for the diagnosis of calicivirus infections in the cat. Vet. Microbiol., 105: 1-7.
CrossRefDirect Link - Reed, L.J. and H. Muench, 1938. A simple method of estimating fifty percent endpoints. Am. J. Epidemiol., 27: 493-497.
CrossRefDirect Link - Schweiger, B., I. Zadow, R. Heckler, H. Timm and G. Pauli, 2000. Application of a fluorogenic PCR assay for typing and subtyping of influenza viruses in respiratory samples. J. Clin. Microbiol., 38: 1552-1558.
Direct Link