
Suzan Bakr Abdu
Department of Biological Sciences, Faculty of Science, King Abdulaziz University, P.O. Box 80200, 21589 Jeddah, Saudi Arabia
LiveDNA: 966.16837
Fatima Motlag Al-Bogami
Department of Biological Sciences, Faculty of Science, King Abdulaziz University, P.O. Box 80200, 21589 Jeddah, Saudi Arabia
International Journal of Pharmacology
Year: 2018 | Volume: 14 | Issue: 5 | Page No.: 717-726







ABSTRACT
Background and Objective: Liver fibrosis is a significant health problem developed as a response to a wound-healing process in injured liver characterized by excessive deposition of fibers and extracellular matrix (ECM). This study aimed to clarify the ultra-structural events that govern the ECM deposition and fibrosis progression using dimethylnitrosamine (DMN) induced liver fibrosis in rat's model. Materials and Methods: Two groups of male rats were assigned, control and DMN. To induce liver fibrosis, rats were administered DMN intraperitoneally (10 mg kg–1, 3 days/week for 21 days). Statistical analysis of animal weights was performed by one-way analysis of variance (ANOVA) and liver tissue was processed for electron microscopy examination. Results: Administration of DMN induced significant body weight loss and severe pathological alterations. The hepatocytes went through apoptosis, the sinusoidal endothelial cells lost their fenestrae and the quiescent hepatic stellate cells (HSCs) were activated, they lost their retinoid, acquired large nucleus and attained large amount of fibers. In other words, HSCs were transformed into myofibroblasts (MFs) phenotype which synthesized ECM proteins and produced fibrous scar. Furthermore, portal fibroblast (PFs) proliferated and produced large amount of fibers in portal and periportal area. Lymphocytic infiltration, necrosis and cholangiocyte proliferation were contributed to liver fibrosis in this study. The most distinctive features of the cellular events of hepatic fibrosis in this study were extensive deposition of ECM and collagens, primarily in portal and periportal areas as well as massive fibrous appearance of the mitochondria of the hepatocytes. Conclusion: Activated portal fibroblasts contributed highly to fibrosis in this study.
PDF Abstract XML References Citation
Received: October 26, 2017;
Accepted: January 03, 2018;
Published: June 15, 2018
Copyright: © 2018. This is an open access article distributed under the terms of the creative commons attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
How to cite this article
Suzan Bakr Abdu and Fatima Motlag Al-Bogami, 2018. Portal Fibroblast Role in Liver Fibrosis in Rats. International Journal of Pharmacology, 14: 717-726.
DOI: 10.3923/ijp.2018.717.726
URL: https://scialert.net/abstract/?doi=ijp.2018.717.726
DOI: 10.3923/ijp.2018.717.726
URL: https://scialert.net/abstract/?doi=ijp.2018.717.726
INTRODUCTION
Liver fibrosis and cirrhosis has become one of the most severe health problems around the world. Liver fibrosis is a reversible wound healing reaction to acute or chronic hepatic injury. During acute injury, the changes in liver architecture are reversible. However, with chronic injury, there is a progressive replacement of the liver parenchyma by scar tissue which can be irreversible1,2.
Liver fibrosis results from the collaboration of different cell types. HSCs are the main initiators of fibrogenesis, coordinating the deposition of ECM in both normal and fibrotic liver2-4 and contributing to approximately 90% of ECM3. HSCs are perisinusoidal cells located in Disse spaces between hepatocytes and sinusoidal endothelial cells3. Moreover, HSCs receive a wide range of signals mediated by cytokines from injured hepatocytes and neighboring cells such as, Kupffer cell, lymphocytes and sinusoidal endothelial cells (SECs), resulting in fibrogenesis3-7.
Previous studies have revealed that during liver injury HSCs become activated to myofibroblasts and proliferate secreting fibrillar collagens, elastin and matrix proteins8,9. When the HSC is activated, it loses its retinoid and starts expressing new receptors such as the platelet derived growth factor (PDGF) receptor and transforming growth factor-β(TGF-β) receptor. It also expresses new proteins such as α-smooth muscle actin, which predominates within vascular smooth muscle cells and plays an important role in fibrogenesis8,10. Myofibroblasts have a crucial role in the development of fibrogenesis. The myofibroblasts proliferate and synthesize large amounts of extracellular component proteins and have the ability to speed up wound repair by contracting the edges of the wound10. These cells have the ability to contract and may contribute to portal hypertension8.
Activation of the HSC into a myofibroblast can be provoked by a range of chronic injuries to the liver such as viral hepatitis, toxins, [non-] alcoholic steatohepatitis and autoimmune disorders11. Previous studies have described fibrosis in two stages activation and resolution. Activation involves two phases, initiation and perpetuation, followed by a resolution phase if the injury recedes1,2. Initiation describes the pre-inflammatory stage. The first paracrine stimulation, such as exposure to lipid peroxides and cytokines from damaged neighboring cells. Once the cell is primed for activation, perpetuation results to maintain an activated phenotype and generate fibrosis2. Activation involves at least seven discrete changes in cell behavior: Proliferation, chemotaxis, fibrogenesis, contractility, matrix degradation, retinoid loss and WBC chemoattractant/cytokine release1,2.
Kupffer cells also contribute to stellate cell activation through stimulating matrix synthesis, cell proliferation and release of retinoid by stellate cells through the actions of cytokines (especially TGF-β) and reactive oxygen species12.
Endothelial cells are also likely to participate in conversion of TGF-β from the latent to active, profibrogenic form. Accordingly, the HSC remodels the ECM into one rich fibril collagens, particularly types I and III. Subsequently, the ECM components increase liver stiffness13. Platelets provide additional paracrine stimulation by PDGF, TGF-β14.
In normal liver, the basement membrane-like matrix of the space of Disse is comprised primarily of collagens IV and VI, which is replaced gradually by collagens I and III and cellular fibronectin during fibrogenesis15. Furthermore, this accumulation of fibrillar ECM is associated with capillarization of the sinusoids, displayed by a loss of the sinusoidal endothelial fenestrae16. Eventually, the accumulation of collagens increases until vascular structures are linked and the architecture of the liver is disrupted significantly17.
A recent study was done by Seki and Schwabe6 has reviewed the mechanisms that link inflammation with the development of liver fibrosis, focusing on the role of inflammatory mediators in HSC activation during fibrosis progression and regression. The study has summarized the contributions of different inflammatory cells as well as cytokines, chemokines and damage associated molecular patterns.
Brenner8 has mentioned that hepatic fibrosis is strongly associated with oxidative stress, increased transforming growth factor-β, hepatocyte death and chronic inflammation. Another study18 suggested that hepatocytes are potent source of reactive oxygen species, generated by membrane injury and lipid peroxidation. In addition, Canbay et al.19 reported that hepatocyte apoptosis following injury promotes the initiation phase of HSC activation.
The study of Nakamura et al.20 and Brenner8 have reported that transforming growth factor-β is the most potent fibrogenic cytokine and plays a critical role in the progression of liver fibrosis.
The last stage is resolution of fibrosis which refers to pathways that either drives the HSC to apoptosis or reversion to a quiescent phenotype21. The aim of this study was to capture the main ultrastructural evidences and advances in liver fibrosis and understanding the mechanisms to explore the different views for therapy.
MATERIALS AND METHODS
Chemicals: N-Nitrosodimethylamine (dimethyl n-nitrosamine, DMN) Cat No: 591068 N-Nitrosodimethylamine-d 6,98 atom% d Sigma (St. Louis, MO, USA).
Experimental animals: Fourteen male Wistar albino rats weighing (90-116 g) were used in the experiment in accordance with the guidelines of the Biochemical and Research Ethical Committee at King Abdulaziz University, Jeddah, Saudi Arabia. This study was done at the School of Pharmacy of King Abdulaziz University in Jeddah, SA. (2017).
Animals were housed in a well-ventilated temperature-controlled room at 22±23°C with 12 h light and dark cycles. Food consisted of standard laboratory rat chow with free access to water. All experimental procedures were performed between 08:00-11:00 am and care was taken to avoid all stressful conditions.
Study design:
| Group 1: | Control treated orally with saline, for 3 weeks |
| Group 2: | DMN treated (10 mg kg–1 b.wt./day) for 3 weeks (3 days/week) |
Ten microliter DMN diluted to 1 mL with 0.15 M sterile NaCl. It was given intraperitoneally (i.p.) on 3 consecutive days of each week for 3 successive weeks22.
The morphological and behavioral changes were monitored after administration of DMN. Animals were weighed on 0, 7, 14, 21 days. Animals were anaesthetized and sacrificed on 21st day from the beginning of the experiment. Sample collection and preparation.
Groups were sacrificed at the end of the3rd week, under ether anesthesia. The livers of all animals were rapidly removed rinsed in cold saline. The liver specimens were fixed in 2.5% glutaraldehyde for electron microscopy study23.
Statistical analysis:
Values for weight were expressed as mean value±standard error. Statistical analysis was performed by one-way analysis of variance (ANOVA) using SPSS 16.0 software (SPSS Inc., USA). Statistical significance was estimated by t-test. The p<0.05 was considered statistically significant.
RESULTS
Body and liver weights: Administration of DMN for 3 weeks has caused a significant decrease in body weight (160.60±8.38) compared to the control (188.20±18.55). The ratio of the total increase in body weight in DMN treated group was significantly low (44.9%) compared to the control group (88.5%) (Table 1). However, the relative liver weight in DMN treated rats was (5.8%) compared to the control (5.2%).
Electron microscopy observations
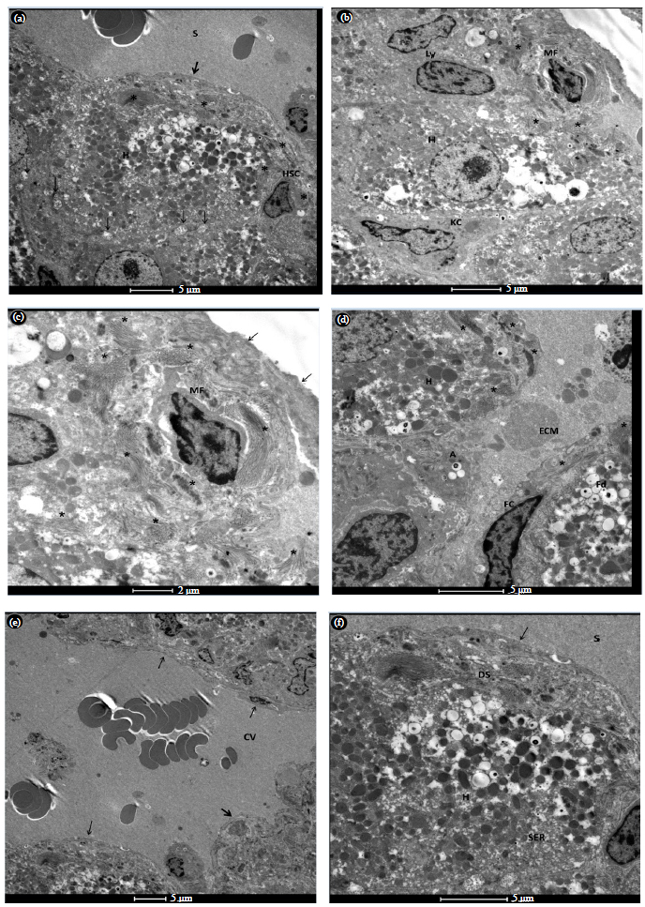
Ultrastructure study: In control liver (G1), the hepatocyte is polygonal two sides of each face hepatic sinusoids and two sides face bile canaliculi. Small irregular microvilli project into the space of Disse from the basal surface of the hepatocytes. Two types of hepatocytes with rough (RER) and smooth (SER) endoplasmic reticulum, dense mitochondria, few lipid droplets, lysosomes and peroxisomes were seen, cells with high density cytoplasm filled with glycogen and cells with low density cytoplasm (Fig. 1a, b). The space of Disse separates hepatocytes from the fenestrated endothelium. This space contains a basement membrane-like matrix which is not electron dense. Hepatic stellate cells are located in the space of Disse with variable size lipid droplets (Fig. 1c). These cells are the primary storage site of retinoid (vitamin A).
| Table 1: | Body and liver weight of the groups treated for 21 days |
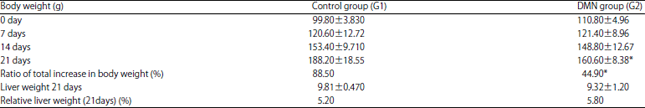 | |
Statistical analysis of rats body weights and liver weights during DMN intoxication. Body and liver weight were measured weekly throughout the study. Results are analyzed by one way ANOVA and are presented as mean±standard error, p<0.05. *Indicates a significant difference between rats treated with DMN and the control | |
 | |
| Fig. 1(a-c): | Transmission electron micrographs of control group (G1) showing (a) Several normal hepatocytes (H), with high and low density cytoplasm, (b) Normal hepatocytes (H) with large number of mitochondria (M) and accumulations of glycogen (Gly). Bile canaliculi (arrows) are sealed off by tight junctions to prevent bile from entering the sinusoids. Note hepatic stellate cell (HSC) in Disse space with fat droplets, Kupffer cell (KC), lysosomes (L) and (c) Enlargement from the previous figure shows hepatic stellate cell (HSC), endothelial cell with fenestrations (arrows), rough endoplasmic reticulum (RER), mitochondria (M), peroxisomes (P) |
Stellate cells in normal liver have spindle-shaped cell bodies with oval or elongated nuclei. The nuclei of hepatic stellate cells were heterochromatic and indented by fat droplets. In control group, the stellate cells were in the quiescent (inactive) state.
Administration of DMN (G2) caused liver damage. Upon liver injury, stellate cells lost their lipid droplets and became activated together with Kupffer cells. Hepatocytes lost their microvilli and endothelial cells lost their characteristic fenestrae (Fig. 2a-f).
Activation of the hepatic stellate cell is characterized by the change of quiescent vitamin A rich cells into fibrogenic and contractile myofibroblast with large nucleus and a thin rim of cytoplasm neighbored by several bundles of collagen fibers (Fig. 2a-c). Coarse collagen fibers in the space of Disse and the portal area were also observed. The sinusoids became dense and fibrous by excessive deposition of ECM and collagen fibers. The perisinusoidal and perivenular hepatocytes were separated from blood flow by collagenous septa (Fig. 2d and e). Many hepatocytes were apoptotic with fibrogenic fragments released from them (Fig. 2d). Hepatocytes were degenerated with numerous fat droplets, in variable sizes and autophagosomes, extensive short arrays of RER, vesiculated SER as well as lysosomes, peroxisomes.
The activated Kupffer cell, were numerous attached to the luminal surface of the sinusoidal endothelium with irregular projections, variable sized vacuoles and a number of lysosomes and phagolysosomes (Fig. 2b).
Capillarization of the hepatocyte endothelium was characterized by the loss of hepatocyte microvilli and the disappearance of endothelial fenestrations (Fig. 2f).
Bile ductules were extended in many areas (Fig. 2g). Nuclei of different cell types were necrotic in many areas in this group (Fig. 2h).
The portal tract was fibrous accompanied by cholangiocyte proliferation. Dramatic changes in nuclear shape and size of the bile ducts. Many ducts were obstructed with distorted microvilli. Portal fibroblasts (PFs) were resided underneath bile ducts, which were surrounded by large amount of collagen fibers.
The most affected areas in the liver tissue were the portal and periportal areas, revealing fibrotic portal space with excessive deposition of ECM proteins and fibrous patches.
 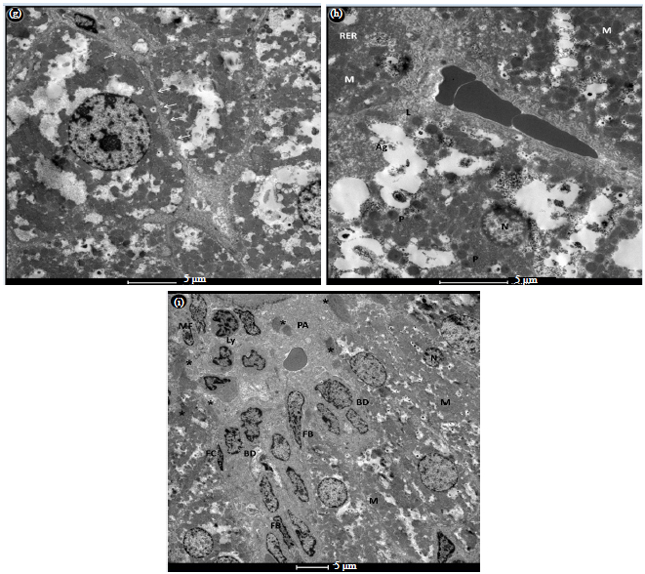 | |
| Fig. 2(a-i): | Transmission electron micrographs of DMN treated group (G2) shows (a) Activated hepatic stellate cell (HSC) with reduced vitamin A content, surrounded by fibers (*). The hepatocytes lost their microvilli (H) and the endothelial cells lost their fenestrae (thick arrow). Proliferated bile ductules many have deteriorated microvilli (arrows). Sinusoid(S), (b) Hepatic stellate cell trans-differentiated from a quiescent vitamin A-rich cell to a myofibroblast (MF) with large nucleus and lost retinoid droplets, surrounded by large amount of fibers (*) in Disse space. Hepatocytes (H) acquired variable sizes of lipid droplets, sinusoid (S). Kupffer cell (K) with lysosomes and phagolysosome. Lymphocytic infiltration (ly), (c) Enlargement of the above micrograph, with large amount of fibers (*) surrounds the myofibroblast (MF), hepatocytes lost their microvilli (H) and plasma membrane definition, defenestrated thickened endothelium (arrows), (d) Apoptotic hepatocyte (H) with large amount of fibers (*) surrounds it. Fibrogenic apoptotic fragment (A), the dense fibrillar (ECM) in the sinusoid. Fat droplets (Fd) filled the upper part of the hepatocyte (thick arrow). Fibrocyte (FC), (e) Fibroblasts surrounding the central vein (second layer cells). Separation of hepatocytes from blood flow by thick collagenous septa (thick arrows). Necrotic defenestrated endothelial cell (arrows), (f) Enlargement of Fig. 2d displaying capillarization, loss of hepatocyte microvilli H) and endothelial cell fenestrations (arrow), fibrous Dissie space (DS) and sinusoid (S), smooth endoplasmic reticulum (SER), (g) Degenerated hepatocytes with extended bile ductules (arrows), (h) Degenerated hepatocyte (H) with extensive short arrays of (RER), dense and ill-defined mitochondria (M), lysosomes (L), Peroxisomes (P). Autophagosomes (AP) and (i) Fibrous portal area (PA) with dense ECM, abnormal nuclear shape and partially obstructed bile duct (BD). The large amount of fibrous patches (*) inside the portal space and the clumps of fibrous undefined mitochondria (M) in the hepatocytes. Necrotic nucleus (N). Fibroblasts (FB) reside underneath of the bile duct epithelium, Fibrocyte (FC). Bile duct (BD) proliferation and lymphocytes infiltration (Ly) |
Most of the hepatocytes became fibrous. The mitochondria were abnormally spread and acquired fibrous appearance.
DISCUSSION
The ultrastructural study has added greatly to researchers knowledge of cellular structure and mechanism of fibrosis. The present study, describes the cellular pathological events of liver fibrogenesis in rats administered DMN. The pathological hallmarks of liver fibrosis in this study are, the hepatocytes were injured and went through apoptosis before fibrosis. The quiescent stellate cells were activated and produced extracellular matrix (ECM) proteins. The sinusoidal endothelial cells (SECs) lost their fenestrae which is called capillarization of the sinusoids. The macrophages of the liver, the Kupffer cells, activated and proliferated and the lymphocytes infiltrated the injured liver and contributed to the inflammation. Moreover, the mitochondria particularly in the portal area were coagulated in abnormal fibrous appearance and become an important indicator of liver fibrosis.
At a cellular level the perisinusoidal hepatic stellate cell (HSC) has been extensively studied as a key effector of fibrogenesis4,21,24.
In the present study, activation of HSC was evidenced by transforming into myofibroblast phenotype cell, that is contractile and fibrogenic, shed its retinoid droplets and produced extraordinary amounts of collagen fibers and fibrotic matrix deposition. Fibers were located around the activated cells in Disse space as previous studies8,25 and also fibers were scattered in the hepatocytes. Collagen and other extracellular matrix (ECM) components are deposited as a response to liver wound-healing process2.
The HSC projections serve a vital role as the cell’s leading edge in "sensing" chemotactic signals to generate a contractile force26. This intimate contact between stellate cells and their neighboring cell types may facilitate intercellular transport of cytokines.
In the present study, bundles of collagens of periportal fibroblasts were prominent. Activated myofibroblasts that derived from perisinusoidal hepatic stellate cells and portal or central vein fibroblasts were proliferated and produced excess extracellular matrix (ECM)27 which led to fibrous portal tract expansion.
Many studies have reviewed the origin of hepatic myofibroblasts which are mainly originate from hepatic stellate cells4,11,25 or from fibroblasts of portal areas25,28,29 or from mesenchymal stem cells or circulating fibrocytes30,31 or through a process of epithelial to mesenchymal transition (EMT) involving either hepatocytes or cholangiocytes32.
Fibrosis results from the interaction of a number of different cell types and results in the buildup of fibrillar collagens as a result of both changes in matrix synthesis and degradation33. Friedman4 reported that the normal subendothelial ECM is critical for maintaining the differentiated functions of resident liver cells. Changes in the microenvironment of the space of Disse (ECM) reflected on all resident liver cells and result in phenotypic changes. In normal liver, ECM is highly dynamic with a balance between synthesis and degradation. However, this balance is disrupted where the ECM becomes gradually insoluble and resistant to protease digestion13. This postulation is in accordance with results in the present study.
In addition, hepatic fibrosis is strongly believed to be associated with oxidative stress, increased transforming growth factor β, hepatocyte death and chronic inflammation8. Oxidative stress generates reactive oxygen species (ROS) which cause liver damage in various ways, such as permanent changes to DNA, lipid peroxidation resulting in destruction of biological membranes and inhibition of mitochondrial and peroxisomal β-oxidation enzymes, leading to accumulation of fatty acids in the hepatocytes, developing hepatic steatosis34. DNA damage, destruction of biological membranes, accumulation of fatty acids in hepatocytes and steatosis, all these manifestations were revealed in the present study as a result of ROS after administration of DMN.
Chronic liver injury causes death of hepatocytes and formation of apoptotic bodies, which release factors that recruit inflammatory cells (neutrophils, monocytes, macrophages and lymphocytes) to the injured liver. Hepatic macrophages (Kupffer cells) produce TGF-β and other inflammatory cytokines that activate collagen type I producing myofibroblasts, which are not present in the normal liver35,36. Hepatic macrophages are often found in close proximity to fibrotic scars9, which suggest it may promote HSC migration9,37. In the present study hepatocytes apoptosis was evident. The apoptotic fragments released from hepatocytes are fibrogenic, as also reported by Canbay et al.19, who linked apoptosis to fibrosis development. Apoptotic hepatocyte bodies have been demonstrated to promote secretion of proinflammatory and fibrogenic cytokines from macrophages and directly promote HSC activation38,39. Moreover, damage-associated molecular patterns (DAMPs) released from dying hepatocytes may also promote fibrogenesis39. In the present study, autophagy was frequently noticed. Autophagy is not only an important cellular degradation mechanism but also plays active role in apoptosis.
Sinusoidal lining endothelial cells SECs represent the safeguard against provoking agents such as toxins and particles delivered to the liver via blood. These cells respond to these conditions by defensive mechanisms called capillarization, loss of fenestrae, to reduce the effect of the toxin on the hepatic cells. Capillarization occurred in chronic liver disease and precedes fibrosis40. The loss of fenestrae prevent hepatocyte oxygenation and have deleterious effect on liver physiology41 increasing resistance to blood flow and may develop portal hypertension42. In the present study, the SECs responded to DMN by thickened and defenestrated endothelium to close the gaps and prevent or reduce exchange between the sinusoid and the space of Disse. In normal liver sinusoid, there is a non-electron dense basement membrane matrix which comprises laminin and type IV collagen. During the development of fibrosis this matrix replaced by one rich in interstitial collagens particularly collagens I and III4,16. This accumulation of fibrillar ECM is associated with a loss of the sinusoidal endothelial fenestrae7,16.
Previous studies have reported that in the chronically injured liver, lymphocytes and HSCs are commonly localized in very close proximity43 as evidenced in the present study suggesting functional interactions.
Cholangiocytes are a rich source of inflammatory cytokines. Bile duct proliferation often results in the activation and expansion of myofibroblasts, surrounding ducts24. A recent study of murine cholestatic liver fibrosis demonstrated that HSCs and not portal fibroblasts are the main ECM-producing cell type. Together, these data suggest that the interaction of cholangiocytes with multiple cell types including lymphocytes, HSCs and portal fibroblasts-contributes to chronic inflammation and fibrosis in cholestatic liver disease24.
Several studies have described antifibrotic effects of platelets14,26. When the HSC is activated, it loses its retinoid and starts expressing new receptors such as the platelet derived growth factor (PDGF) receptor and transforming growth factor (TGF-β) receptor. It also expresses new proteins such as α smooth muscle actin. The activated HSC proliferates and synthesizes extracellular matrix proteins to produce the fibrous scar. The HSCs contract and may contribute to portal hypertension8. Brenner8 suggested that through the production of the metalloproteinase, MMP2, the HSCs contribute to the degradation of the normal extracellular matrix. TGF-β exerts acritical role in the progression of established fibrosis.
In the present study, as the liver becomes fibrotic, the total content of collagens components increases, accompanied by a shift in the type of ECM in the subendothelial space sinusoids from the normal low-density to high-density ECM. The extensive short and thick arrays of rough endoplasmic reticulum in hepatocytes are indicative of synthetic protein activity.
In the present study, liver resident activated hepatic stellate cells (HSCs) and activated portal fibroblasts (PFs) are the major source of the fibrous scar in the liver as mentioned earlier by Iwaisako et al.29 in PNAS.
CONCLUSION AND FUTURE
RECOMMENDATION
The present study has proved that PFs are major contributor in fibrosis. This study has uncovered the intricate ultrastructural mechanism that occurred during liver fibrogenesis and giving the opportunities to develop targeted therapies to reverse the fibrotic response of chronic liver disease. This study may not only improve understanding of this complex mechanism but also allow one to focus on therapeutic molecules and inhibitors. Authors expect that future research will focus on therapeutic strategies.
SIGNIFICANCE STATEMENT
This study discovers the role of portal fibroblast that can be beneficial for liver fibrosis induced rats. This study will help the researchers to uncover the critical areas of the origin of myofibroblasts and their role in pathological wound healing that many researchers were not able to explore. Thus a new theory on the contribution of portal fibroblasts to liver fibrosis may be arrived at.
ACKNOWLEDGMENTS
This study was supported by a grant from King Abdulaziz city for science and technology, Riyadh, Saudi Arabia. No. 58-37. The authors appreciate the technical assistance of the team of the Electron Microscopy Unit at KAUST University in Saudi Arabia.
REFERENCES
- Lee, U.E. and S.L. Friedman, 2011. Mechanisms of hepatic fibrogenesis. Best Pract. Res. Clin. Gastroenterol., 25: 195-206.
CrossRefDirect Link - Pradere, J.P., J. Kluwe, S. de Minicis, J.J. Jiao and G.Y. Gwak et al., 2013. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology, 58: 1461-1473.
CrossRefDirect Link - Friedman, S.L., 2008. Hepatic stellate cells: Protean, multifunctional and enigmatic cells of the liver. Physiol. Rev., 88: 125-172.
CrossRefDirect Link - Ding, B.S., Z. Cao, R. Lis, D.J. Nolan and P. Guo et al., 2014. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature, 505: 97-102.
CrossRefDirect Link - Seki, E. and R.F. Schwabe, 2015. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology, 61: 1066-1079.
CrossRefDirect Link - Nafady, A.M., O.B. Ahmed and H.H. Ghafeer, 2017. Scanning and transmission electron microscopy of the cells forming the hepatic sinusoidal wall of rat in acetaminophen- and Escherichia coli endotoxin-induced hepatotoxicity. J. Microsc. Ultrastruc., 5: 21-27.
CrossRefDirect Link - Brenner, D.A., 2009. Molecular pathogenesis of liver fibrosis. Trans. Am. Clin. Climatol. Assoc., 120: 361-368.
Direct Link - Pellicoro, A., R.L. Aucott, P. Ramachandran, A.J. Robson and J.A. Fallowfield et al., 2012. Elastin accumulation is regulated at the level of degradation by macrophage metalloelastase (MMP-12) during experimental liver fibrosis. Hepatology, 55: 1965-1975.
CrossRefDirect Link - Bataller, R. and D.A. Brenner, 2005. Liver fibrosis. J. Clin. Invent., 115: 209-218.
CrossRefPubMedDirect Link - Bilzer, M., F. Roggel and A.L. Gerbes, 2006. Role of kupffer cells in host defense and liver disease. Liver Int., 26: 1175-1186.
CrossRefDirect Link - Schuppan, D., M. Ruehl, R. Somasundaram and E.G. Hahn, 2001. Matrix as a modulator of hepatic fibrogenesis. Semin. liver Dis., 21: 351-372.
CrossRefPubMedDirect Link - Takahashi, K., S. Murata, K. Fukunaga and N. Ohkohchi, 2013. Human platelets inhibit liver fibrosis in severe combined immunodeficiency mice. World J. Gastroenterol., 19: 5250-5260.
CrossRefDirect Link - Brown, B., K. Lindberg, J. Reing, D.B. Stolz and S.F. Badylak, 2006. The basement membrane component of biologic scaffolds derived from extracellular matrix. Tissue Eng., 12: 519-526.
CrossRefDirect Link - Friedman, S.L., F.J. Roll, J. Boyles and D.M. Bissell, 1985. Hepatic lipocytes: The principal collagen-producing cells of normal rat liver. Proc. Natl. Acad. Sci. USA., 82: 8681-8685.
Direct Link - Issa, R., X. Zhou, C.M. Constandinou, J. Fallowfield and H. Millward-Sadler et al., 2004. Spontaneous recovery from micronodular cirrhosis: Evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology, 126: 1795-1808.
CrossRefDirect Link - Novo, E., F. Marra, E. Zamara, L. Valfre di Bonzo and A. Caligiuri et al., 2006. Dose dependent and divergent effects of superoxide anion on cell death, proliferation and migration of activated human hepatic stellate cells. Gut, 55: 90-97.
CrossRefPubMedDirect Link - Canbay, A., S. Friedman and G.J. Gores,, 2004. Apoptosis: The nexus of liver injury and fibrosis. Hepatology, 39: 273-278.
Direct Link - Nakamura, T., R. Sakata, T. Ueno, M. Sata and H. Ueno, 2000. Inhibition of transforming growth factor β prevents progression of liver fibrosis and enhances hepatocyte regeneration in dimethylnitrosamine‐treated rats. Hepatology, 32: 247-255.
CrossRefDirect Link - Friedman, S.L., 2012. Fibrogenic cell reversion underlies fibrosis regression in liver. Proc. Natl. Acad. Sci. USA., 109: 9230-9231.
CrossRefDirect Link - Lee, E.S., H.E. Lee, J.Y. Shin, S. Yoon and J.O. Moon, 2003. The flavonoid quercetin inhibits dimethylnitrosamine-induced liver damage in rats. J. Pharm. Pharmacol., 55: 1169-1174.
CrossRefDirect Link - Mederacke, I., C.C. Hsu, J.S. Troeger, P. Huebener and X. Mu et al., 2013. Fate-tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its etiology. Nat. Commun., Vol. 4.
CrossRefDirect Link - Parola, M., F. Marra and M. Pinzani, 2008. Myofibroblast-like cells and liver fibrogenesis: Emerging concepts in a rapidly moving scenario. Mol. Aspects Med., 29: 58-66.
CrossRefDirect Link - Melton, A.C. and H.F. Yee Jr., 2007. Hepatic stellate cell protrusions couple platelet‐derived growth factor‐bb to chemotaxis. Hepatology, 45: 1446-1453.
CrossRefDirect Link - Xu, J., X. Liu, Y. Koyama, P. Wang and T. Lan et al., 2014. The types of hepatic myofibroblasts contributing to liver fibrosis of different etiologies. Front. Pharmacol., Vol. 5.
CrossRefDirect Link - Dranoff, J.A. and R.G. Wells, 2010. Portal fibroblasts: Underappreciated mediators of biliary fibrosis. Hepatology, 51: 1438-1444.
CrossRefDirect Link - Iwaisako, K., C. Jiang, M. Zhang, M. Cong and T.J. Moore-Morris et al., 2014. Origin of myofibroblasts in the fibrotic liver in mice. Proc. Natl. Acad. Sci. USA., 111: E3297-E3305.
CrossRefDirect Link - Kisseleva, T. and D.A. Brenner, 2008. Mechanisms of fibrogenesis. Exp. Biol. Med., 233: 109-122.
CrossRefDirect Link - Henderson, N.C. and S.J. Forbes, 2008. Hepatic fibrogenesis: From within and outwith. Toxicology, 254: 130-135.
CrossRefDirect Link - Cannito, S., E. Novo, L.V. di Bonzo, C. Busletta, S. Colombatto and M. Parola, 2010. Epithelial-mesenchymal transition: From molecular mechanisms, redox regulation to implications in human health and disease. Antioxid. Redox Signal., 12: 1383-1430.
CrossRefDirect Link - Iredale, J.P., A. Thompson and N.C. Henderson, 2013. Extracellular matrix degradation in liver fibrosis: Biochemistry and regulation. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis., 1832: 876-883.
CrossRefDirect Link - Reddy, J.K. and M.S. Rao, 2006. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am. J. Physiol. Gastrointest. Liver Physiol., 290: G852-G858.
CrossRefPubMedDirect Link - Kisseleva, T., M. Cong, Y. Paik, D. Scholten and C. Jiang et al., 2012. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. USA., 109: 9448-9453.
CrossRefDirect Link - Ahmad, A. and R. Ahmad, 2014. Resveratrol mitigate structural changes and hepatic stellate cell activation in N'-nitrosodimethylamine-induced liver fibrosis via restraining oxidative damage. Chemico-Biol. Interact., 221: 1-12.
CrossRefDirect Link - Marra, F. and F. Tacke, 2014. Roles for chemokines in liver disease. Gastroenterology, 147: 577-594.e1.
CrossRefDirect Link - Ali, C., E. Ariel, H. Feldstein, N. Higuchi, A. Werneburg, S. Grambihler and J. Gregory, 2003. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology, 38: 1188-1198.
CrossRefDirect Link - Luedde, T., N. Kaplowitz and R.F. Schwabe, 2014. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology, 147: 765-783.e4.
CrossRefDirect Link - Xie, G., L. Wang, X. Wang and L.D. DeLeve, 2010. Isolation of periportal, midlobular and centrilobular rat liver sinusoidal endothelial cells enables study of zonated drug toxicity. Am. J. Physiol. Gastrointest. Liver Physiol., 299: 1204-1210.
CrossRefDirect Link - Le Couteur, D.G., V.C. Cogger, A. Markus, P.J. Harvey, Z.L. Yin, A.D. Ansselin and A.J. McLean, 2001. Pseudocapillarization and associated energy limitation in the aged rat liver. Hepatology, 33: 537-543.
CrossRefDirect Link - Xu, B., U. Broome, M. Uzunel, S. Nava and X. Ge et al., 2003. Capillarization of hepatic sinusoid by liver endothelial cell-reactive autoantibodies in patients with cirrhosis and chronic hepatitis. Am. J. Pathol., 163: 1275-1289.
CrossRefDirect Link - Muhanna, N., A. Horani, S. Doron and R. Safadi, 2007. Lymphocyte-hepatic stellate cell proximity suggests a direct interaction. Clin. Exp. Immunol., 148: 338-347.
CrossRefDirect Link