
Xiaokang Wang
Department of Pharmacy, Shenzhen Longhua District Central Hospital, 518110 Shenzhen, China
Caiyun Jiang
Department of Pharmacy, The Third Affiliated Hospital of Sun Yat-Sen University, 510630 Guangzhou, China
Xiaosong Wu
Department of Pharmacy, The Second Xiangya Hospital, Central South University, Changsha, Hunan, 410011, China
Peng Zou
Department of Anesthesiology, The First Affiliated Hospital of Jinan University, 510630 Guangzhou, China
LiveDNA: 86.16520
Zhufeng Wu
Department of Pharmacy, The Second Xiangya Hospital, Changsha, China
LiveDNA: 86.16523
International Journal of Pharmacology
Year: 2018 | Volume: 14 | Issue: 3 | Page No.: 320-328







ABSTRACT
Background and Objective: UDP-glucuronosyltransferases 1A8 (UGT1A8) is an important enzyme responsible for glucuronidation of numerous xenobiotic/drugs. The objective of this study was to establish a substrate selectivity model through pharmacophore approach. Methodology: Thirty-six substrates of UGT1A8 collected from the literature were divided into training (n = 24) and test sets (n = 12). The Discovery Studio 2.5 (DS) software was utilized to establish the pharmacophore model. The HypoGen algorithm that was available in 3D QSAR Pharmacophore Generation protocol was applied to construct pharmacophore hypotheses. Correlation analyses were performed between the predicted activity and the experimental activity of the training and test sets. Results: The established pharmacophore model consisted of 2 hydrogen-bonding acceptors and one aromatic ring. The best pharmacophore model (hypothesis 1) was statistically significant with high value of correlation coefficient and low value of difference between the null cost and the total cost. Besides, the predicted catalysis activities were within one log residual of experimental value for substrates in the test set. Conclusion: Pharmacophore model for UGT1A8 was successfully constructed for the first time in this study. The established model contributed to an improved understanding of the UGT1A8’s substrate selectivity. Besides, this model would be an efficient tool for high-throughput prediction of UGT1A8 metabolism.
PDF Abstract XML References Citation
Received: June 08, 2017;
Accepted: September 25, 2017;
Published: March 15, 2018
Copyright: © 2018. This is an open access article distributed under the terms of the creative commons attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
How to cite this article
Xiaokang Wang, Caiyun Jiang, Xiaosong Wu, Peng Zou and Zhufeng Wu, 2018. Substrate Selectivity for UDP-glucuronosyltransferase1A8 using the Pharmacophore Approach. International Journal of Pharmacology, 14: 320-328.
DOI: 10.3923/ijp.2018.320.328
URL: https://scialert.net/abstract/?doi=ijp.2018.320.328
DOI: 10.3923/ijp.2018.320.328
URL: https://scialert.net/abstract/?doi=ijp.2018.320.328
INTRODUCTION
Glucuronidation is a major pathway for xenobiotic/drugs metabolism and excretion in humans and other mammalian species1. This reaction occurs via transferring a glucuronic acid from cofactor UDP-glucuronic acid (UDPGA) to a compound which usually contains hydroxyl, carboxyl or nitrogen group2. Glucuronidation can enhance hydrophilicity of a compound, resulting in enhanced excretion rate of xenobiotic/drugs from the body1. In most instances, the glucuronidated metabolites are inactive3. However, glucuronidation can generate pharmacologic or toxicologic activity in some instances3. For example, morphine-6-glucuronide is a more potent opioid agonist than morphine4. On the basis of amino acid sequence identity, UDP-glucuronosyltransferases (UGTs), which specifically catalyze the reaction of glucuronidation, are divided into four families: UGT1, UGT2, UGT3 and UGT85. The most important drug-conjugating UGTs are UGT1A and UGT2B family1. Of note, UGT 1A8 expressed in the small intestine and colon plays a key role in clearance of many compounds such as isoflavones, gingerols and curcumin ana logs6-8.
Pharmacophore model is a collectivity of electronic and steric features which is essential for the optimal supramolecular interaction with a specific biologic target to initiate (or block) its biologic response9. There are 2 kinds of pharmacophore model, namely, ligand-based and structure-based pharmacophore model10. Ligand-based pharmacophore model is constructed by overlying a group of active compounds and extracting common chemical features which are necessary for their bioactivity. On the contrary, structure-based pharmacophore model can only be utilized when a three dimensional (3D) structure of a biological target is available. Besides, pharmacophore techniques have been widely used as a 3D-QSAR (3D-Quantitative structure-activity relationship) method to predict ADME (Absorption, Distribution, Metabolism and Excretion) properties in recent years11. The substrate selectivity of many UGTs (e.g., UGT1A1, UGT1A3, UGT1A7) and cytochrome P450 enzymes involved in the oxidation of much xenobiotic/drugs (e.g., CYP 1A2, 2B6, 2C9 and 3A4) have been well understood and predicted using pharmacophore approach11,12.
High rate of drug attritions usually caused by the poor properties of ADME13. Hence, it is necessary to deal with ADME issues earlier in the drug discovery cycle using effective computational tools. However, QSAR models are still unavailable for UGT1A8 enzyme. Therefore, a predictive QSAR model for UGT1A8 is of great value in an attempt to predict the glucuronidation of drug candidates. In this study, we have evaluated the potential of pharmacophore approach in quantifying the substrate selectivity of UGT1A8. Discovery Studio 2.5 was used to build the pharmacophore model. Thirty-six UGT1A8’s substrates collated from the literature were divided into two sets, namely, training set (n = 24) and test set (n = 12). The predictive power of the established pharmacophore model was validated via cost analysis, Fisher’s randomization test and test set verification. It was demonstrated for the first time that the pharmacophore model possessed predictive capability was successfully constructed for UGT1A8, contributing an improved understanding of the UGT1A8’s substrate selectivity and a more comprehensive prediction of UGT-mediated metabolism.
MATERIALS AND METHODS
Data preparation: For the pharmacophore modeling study, (Fig. 1) a series of thirty-six diverse substrates of UGT1A8 were collected from the literature at Jinan University in 2016. The intrinsic clearance value (CLint) that derived from kinetic determination was regarded as glucuronidation activity. The CLint values and references for all substrates were listed at Table 1 and 2. Based on the principles of wide coverage of the activity (four orders of magnitude) and chemical structural diversity, all substrates were randomly divided into training (n = 24) and test sets (n = 12).
| Table 1: | Experimental UGT1A8 activities [(CLint or log(CLint)] for the training set compounds |
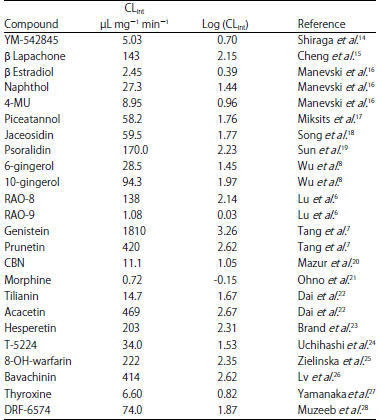 | |
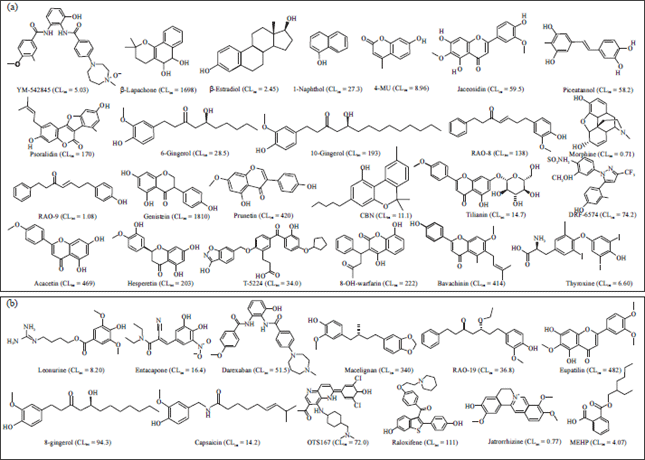 | |
| Fig. 1(a-b): | Chemical structures of the thirty-six UGT1A8 substrates collated from literature. CLint values were included in parentheses. The unit of CLint values is L mg–1 min–1. References are provided in Supplementary materials, (a) Panel 24 UGT1A8 substrates in the training set, (b) Panel 12 UGT1A8 substrates in the test set |
| Table 2: | Experimental UGT 1A8 activities [(CLint or log (CLint)] for the test set compounds |
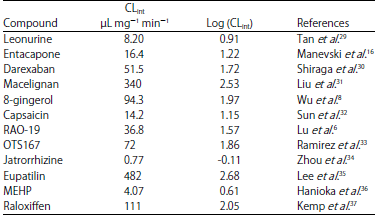 | |
The training set was applied to construct the pharmacophore model, while the test set was served to evaluate the external predictive ability of the established model. The three dimensional (3D) structures of all compounds were prepared by Discovery Studio 2.5 (DS) (Accelrys, US). Gasteiger-Marsili method was used to determine partial atomic charges.
Pharmacophore model generation: The generation of pharmacophore model was performed using Discover Studio 2.5 (DS) (Accelrys, US). Firstly, the Generate Conformation protocol was used to generate conformations for substrates in the training set. During the generation of diverse conformations for each molecule, a maximum of 255 conformations within an energy range of 20 kcal mol–1 was set. "Best" method was implemented for the conformation fitting and generation. All other parameters were kept as their default values. Secondly, the HypoGen algorithm available in 3D QSAR Pharmacophore Generation protocol was utilized to build pharmacophore hypotheses. A set of eleven chemical features including Hydrogen-Bond Acceptor Lipid (HBAL), Hydrogen-Bond Acceptor (HBA), Hydrogen-Bond Donor Lipid (HBDL), Hydrogen-Bond Donor (HBD), aromatic ring (RA), hydrophobic (HY), Hydrophobic Aromatic (HYAr), Hydrophobic Aliphatic (HYAI), positively (PC) and negatively (NC) charged, positively (PI) and negatively (NI) ionisable were available in the HypoGen. Taking into account of the chemical structure of molecules in the training set, 4 pharmacophore features were served as the hypothesis generation: The default HBA, HBD, RA and HY features. A minimum of 1 and a maximum of 5 features in above 4 pharmacophore features were chosen to generate 10 statistically significant model.
Pharmacophore model validation: In this study, the generated model was validated by three methods: Cost analysis, Fisher’s randomization test and the test set prediction38. First, the quality of a pharmacophore model can be assessed in terms of null cost, total cost and fixed cost. As a fine pharmacophore model, the difference between the null cost and the total cost values should be small. Besides, the fixed cost should be close to the total cost. Then, the Fisher’s randomization test was applied to assess the statistical relevance to the model. In this test, the activity values of substrates in the training set were randomly reassigned before hypotheses generation. The confidence level was set to 95%, where 19 random hypotheses were yielded. Finally, the test set prediction was used as an external model validation method. The substrates in the test were mapped to the best pharmacophore model to obtain their predicted CLint values using the Ligand Pharmacophore Mapping protocol39.
Statistical analysis: Correlation analyses were performed between the predicted and experimental activities of the training and test sets. Correlation analyses were performed by GraphPad Prism V5 software.
RESULTS
Construction of the pharmacophore model: Twenty-four UGT1A8 substrates included in the training set (Fig. 1a) were used to construct the pharmacophore model. The activities (CLint) for these compounds spanned 4 orders of magnitude (Fig. 1). Ten pharmacophore hypotheses were generated by the HypoGen algorithm. It was interesting to find that all hypotheses included 2 Hydrogen-Bond Acceptors (HBA) and one aromatic ring (RA), suggesting that these chemical features play an important role in the UGT1A8 activity (Table 3). On the basic of cost and correlation analysis, hypothesis 1was regarded as the best model (Table 3). The structural features and geometry of hypothesis 1 were presented in Fig. 2a. The aromatic ring was 5.2 Å away from one hydrogen-bond acceptor and 4.5 Å away from the other one. The most active substrate genistein (CLint = 1810 μL mg–1 min–1) was well mapped to the model features (Fig. 2b). By contrary, the least active substrate morphine (CLint = 0.71 μL mg–1 min–1) was poorly fitted to all features (Fig. 2c). This suggested that the established pharmacophore model could distinguish good UGT1A8 substrates from poor ones.
Validation of the pharmacophore model: Firstly, cost analysis was applied to model validation. The cost values for all hypotheses were listed at Table 3. The total cost (113.79) was close to the fixed cost (104.65) for hypothesis 1. Besides, the difference between the null cost and the total cost was 43.73, indicating that there was a high chance (75-90%) that the model represented a good correlation of the information. This was agreed well with the fact that this model showed an excellent correlation coefficient (r = 0.8334) between predicted and experimental activities for substrates in training set (Fig. 3). Furthermore, the configuration cost (10.431) was not greater than 17, suggesting that a standard HypoGen algorithm was performed40.
The pharmacophore model was then assessed by cross-validation using Fischer’s randomization method.
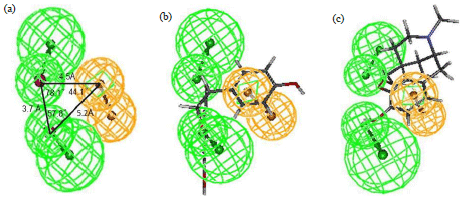 | |
| Fig. 2(a-c): | Best pharmacophore model for UGT1A8., (a) Panel Three dimensional (3D) diagram of the model (Green sphere, HBA, Orange sphere, RA). (b) Panel Mapping of the most active substrate genistein to the model. (c) Panel Mapping of the least active substrate morphine to the model |
| Table 3: | Statistical information of the top ten hypotheses as a result of pharmacophore model generation |
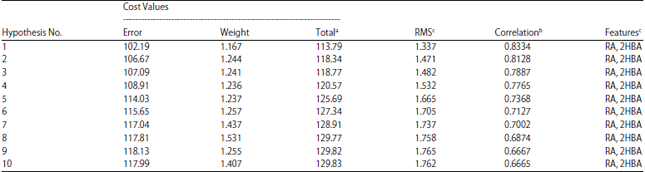 | |
aTotal cost: Configuration cost+error cost+weight cost, where the configuration cost = 10.431, the fixed cost = 104.65 and the null cost = 157.52, bCorrelation coefficient (R) between the predicted activity and the experimental activity of the training set, cAbbreviations used: RMS-Root mean square deviation, HBA-Hydrogen-bond acceptor, RA-Ring aromatic | |
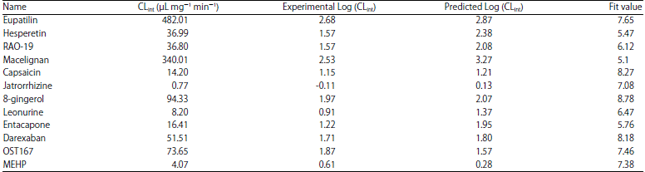
| Table 4: | Experimental and predicted UGT1A8 activities (CLint or log(CLint) values) by pharmacophore model for the substrates in the test set |
 | |
 | |
| Fig. 3: | Correlation analysis between predicted and experimental activities for the training set |
Nineteen-hypotheses were generated randomly (Fig. 4). Clearly, all 19 generated hypotheses after randomization showed higher total cost value compared to the original hypothesis (Fig. 4). This result demonstrated that the original hypothesis was not generated randomly.
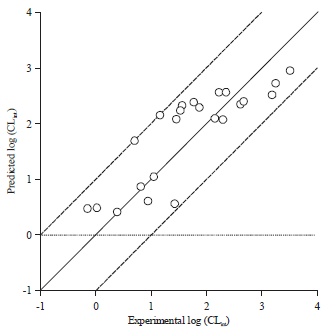
Finally, the correlation coefficient between predicted and experimental values for the test set substrates (Fig. 1b) was used to evaluate reliability of the hypothesis 1.Predicted and experimental values for the test set were listed at Table 4. A good correlation coefficient (r2 = 0.823) was obtained for the model. Besides, the predicted CLint values were close to the experimental values for all substrates, deviating by no more than one log unit (Fig. 5). In fact, predicted value within one log residual of experimental value was considered satisfactory in drug metabolism field41,42.
DISCUSSION
In this study, a 3D QSAR model for UGT 1A8 was successfully established by pharmacophore approach for the first time. The pharmacophore model consisted of two HBA and one RA chemical features, demonstrating that HBA and RA served an important role in the catalyzing capability of UGT 1A8 (Fig. 2). Therefore, this study highlighted an overlay of chemical characteristics related to UGT1A8 substrates.
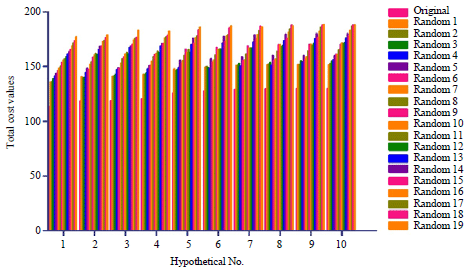 | |
| Fig. 4: | Comparison of randomly generated hypotheses and original hypotheses. The confidence level was set as 95% |
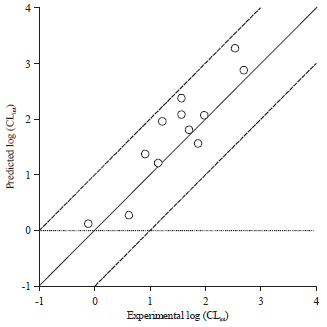 | |
| Fig. 5: | Correlation analysis between predicted and experimental activities for the test set |
Furthermore, this model possessed strong capacity to predict substrate activity for UGT 1A8 (Fig. 4). Accordingly, the established model would be an efficient tool for high-through put prediction of UGT1A8 metabolism. UGT1A8 was a major UGT enzyme contributing to drug metabolism in the gastro intestinal tract3. However, the computational models were still unavailable for UGT 1A8. Hence, construction of the pharmacophore model for UGT1A8 in this study would be helpful for the prediction of UGT-mediated metabolism in the gastrointestinal tract.
Many drug attritions caused by undesirable ADME properties have led to the need to identify ADME problems in the drug discovery process as early as possible43. In fact, there 2 kind approaches (i.e., in silico and in vitro) could beutilized to optimize the selection of the most suitable drug candidates for development42. In silico approaches were effective to deal with ADME problems still earlier in the drug discovery process and helpful to select better drug candidates from many compounds to move forward44. On the contrary, in vitro approaches for ADME issues were time and economy consuming process. Of note, the pharmacophore technique was a powerful computational tool for ADME properties’ prediction, which has successfully quantified substrate selectivity for various CYP and UGT enzymes11.
Because the full crystal structure for UGT 1A8 was unavailable, the ligand-based pharmacophore method was used to build the quantitative pharmacophore model. In order to ensure meaningful performing of the HypoGen algorithm, 24 structurally diverse substrates of UGT 1A8 covering a range of more than 4 log units were selected for model building (Fig. 1). Therefore, it was reasonable that ten pharmacophore models were statistically significant with high values of correlation coefficients and low values of total cost (Table 3). Besides, the hypothesis 1 (the best pharmacophore model) correctly predicted the activities of substrates in the test set (deviations of less than one log unit), suggestive of a good external predicted ability of the model (Fig. 5).
The pharmacophore model suggested that hydrogen-bonding acceptor played a significant role in the molecules glucuronidation by UGT1A8. Hydrogen-bond acceptor was regarded as a hydrophilic group. Accordingly, this result was also supported the fact that hydrophilic region was an important contributor to substrate catalysis. Besides, pharmacophore analysis was firstly demonstrated that ring aromatic was an essential determinant for glucuronidation activity by UGT enzyme. Ring aromatic provided strong hydrophobic properties in molecules. Noteworthily, hydrophobicity has been found to be an important factor for the binding of substrates to UGT enzymes such as UGT1A1, UGT1A4 and UGT1A1012. Because catalysis efficiency was closely related to substrate binding to the enzyme, it was reasonable to find that aromatic ring was important for molecule’s glucuronidation by UGT 1A8.
CONCLUSION
It is concluded, a quantitative model for UGT 1A8 was firstly constructed using pharmacophore approach in the present study. The pharmacophore model was composed of two hydrogen-bonding acceptors and one aromatic ring. The best pharmacophore model (hypothesis 1) was statistically significant with high values of correlation coefficients and low values of difference between the null cost and the total cost. Besides, the established model was able to accurately predicted catalysis activity within one log residual of laboratorial value for substrates in the test set, highlighting the predictability of the model. Application of the established model to predict UGT1A8 metabolism, hence, is fully expected in drug development problems. Our model could also contribute to an improved understanding of the substrate selectivity of UGT1A8.
SIGNIFICANCE STATEMENT
This study discovers the substrate selectivity of UGT1A8 that can be beneficial for high-throughput prediction of UGT1A8 metabolism. This study will help the researchers to uncover the critical areas of computational prediction for metabolism that many researchers were not able to explore.
ACKNOWLEDGMENT
This research was supported by the Shenzhen Longhua New District Science and Technology Innovation Fund Projects with grant No. 20160523A1030149.
REFERENCES
- Wu, B., K. Kulkarni, S. Basu, S. Zhang and M. Hu, 2011. First-pass metabolism via UDP-glucuronosyltransferase: A barrier to oral bioavailability of phenolics. J. Pharm. Sci., 100: 3655-3681.
CrossRefDirect Link - Ritter, J.K., 2000. Roles of glucuronidation and UDP-glucuronosyltransferases in xenobiotic bioactivation reactions. Chemico-Biol. Interact., 129: 171-193.
CrossRefDirect Link - Rowland, A., J.O. Miners and P.I. Mackenzie, 2013. The UDP-glucuronosyltransferases: Their role in drug metabolism and detoxification. Int. J. Biochem. Cell Biol., 45: 1121-1132.
CrossRefDirect Link - Mikus, G. and R. Klimas, 2015. Relative contribution of morphine and morphine-6-glucuronide to the analgesic effect after morphine administration. Br. J. Anaesth., 114: 1007-1007.
CrossRefDirect Link - Mackenzie, P.I., K.W. Bock, B. Burchell, C. Guillemette and S.I. Ikushiro et al., 2005. Nomenclature update for the mammalian UDP Glycosyltransferase (UGT) gene superfamily. Pharmacogenet. Genom., 15: 677-685.
Direct Link - Lu, D., H. Liu, W. Ye, Y. Wang and B. Wu, 2017. Structure- and isoform-specific glucuronidation of six curcumin analogs. Xenobiotica, 47: 304-313.
CrossRefDirect Link - Tang, L., R. Singh, Z. Liu and M. Hu, 2009. Structure and concentration changes affect characterization of UGT isoform-specific metabolism of isoflavones. Mol. Pharm., 6: 1466-1482.
CrossRefDirect Link - Wu, Z., H. Liu and B. Wu, 2015. Regioselective glucuronidation of gingerols by human liver microsomes and expressed UDP-glucuronosyltransferase enzymes: Reaction kinetics and activity correlation analyses for UGT1A9 and UGT2B7. J. Pharm. Pharmacol., 67: 583-596.
CrossRefDirect Link - Buckle, D.R., P.W. Erhardt, C.R. Ganellin, T. Kobayashi, T.J. Perun, J. Proudfoot and J. Senn-Bilfinger, 2013. Glossary of terms used in medicinal chemistry. Part II (IUPAC Recommendations 2013). Pure Applied Chem., 85: 1725-1758.
CrossRefDirect Link - Yang, S.Y., 2010. Pharmacophore modeling and applications in drug discovery: Challenges and recent advances. Drug Discov. Today, 15: 444-450.
CrossRefDirect Link - Guner, O.F. and J.P. Bowen, 2013. Pharmacophore modeling for ADME. Curr. Top. Med. Chem., 13: 1327-1342.
Direct Link - Dong, D., R. Ako, M. Hu and B. Wu, 2012. Understanding substrate selectivity of human UDP-glucuronosyltransferases through QSAR modeling and analysis of homologous enzymes. Xenobiotica, 42: 808-820.
CrossRefDirect Link - Ortega, S.S., L.C. Cara and M.K. Salvador, 2012. In silico pharmacology for a multidisciplinary drug discovery process. Drug Metabol. Drug Interact., 27: 199-207.
CrossRefPubMedDirect Link - Shiraga, T., K. Yajima, T. Teragaki, K. Suzuki and T. Hashimoto et al., 2012. Identification of enzymes responsible for the N-oxidation of darexaban glucuronide, the pharmacologically active metabolite of darexaban and the glucuronidation of darexaban N-oxides in human liver microsomes. Biol. Pharm. Bull., 35: 413-421.
CrossRefDirect Link - Cheng, X., F. Liu, T. Yan, X. Zhou and L. Wu et al., 2012. Metabolic profile, enzyme kinetics and reaction phenotyping of β-lapachone metabolism in human liver and intestine in vitro. Mol. Pharm., 9: 3476-3485.
CrossRefDirect Link - Manevski, N., J. Troberg, P. Svaluto-Moreolo, K. Dziedzic, J. Yli-Kauhaluoma and M. Finel, 2013. Albumin stimulates the activity of the human UDP-glucuronosyltransferases 1A7, 1A8, 1A10, 2A1 and 2B15, but the effects are enzyme and substrate dependent. PLoS ONE, Vol. 8.
CrossRefDirect Link - Miksits, M., A. Maier-Salamon, T.P. Nha Vo, M. Sulyok, R. Schuhmacher, T. Szekeres and W. Jager, 2010. Glucuronidation of piceatannol by human liver microsomes: Major role of UGT1A1, UGT1A8 and UGT1A10. J. Pharm. Pharmacol., 62: 47-54.
CrossRefDirect Link - Song, W.Y., H.Y. Ji, N.I. Baek, T.S. Jeong and H.S. Lee, 2010. In vitro metabolism of jaceosidin and characterization of cytochrome P450 and UDP-glucuronosyltransferase enzymes in human liver microsomes. Arch. Pharmacal Res., 33: 1985-1996.
CrossRefDirect Link - Sun, H., Z. Ma, D. Lu and B. Wu, 2015. Regio- and isoform-specific glucuronidation of psoralidin: Evaluation of 3-O-glucuronidation as a functional marker for UGT1A9. J. Pharm. Sci., 104: 2369-2377.
CrossRefDirect Link - Mazur, A., C.F. Lichti, P.L. Prather, A.K. Zielinska and S.M. Bratton et al., 2009. Characterization of human hepatic and extrahepatic UDP-glucuronosyltransferase enzymes involved in the metabolism of classic cannabinoids. Drug Metab. Dispos., 37: 1496-1504.
CrossRefDirect Link - Ohno, S., K. Kawana and S. Nakajin, 2008. Contribution of UDP-glucuronosyltransferase 1A1 and 1A8 to morphine-6-glucuronidation and its kinetic properties. Drug Metab. Dispos., 36: 688-694.
CrossRefDirect Link - Dai, P., F. Luo, Y. Wang, H. Jiang and L. Wang et al., 2015. Species- and gender-dependent differences in the glucuronidation of a flavonoid glucoside and its aglycone determined using expressed UGT enzymes and microsomes. Biopharmaceut. Drug Dispos., 36: 622-635.
CrossRefDirect Link - Brand, W., M.G. Boersma, H. Bik, E.F. Hoek-van den Hil and J. Vervoort et al., 2010. Phase II metabolism of hesperetin by individual UDP-glucuronosyltransferases and sulfotransferases and rat and human tissue samples. Drug Metab. Dispos., 38: 617-625.
CrossRefDirect Link - Uchihashi, S., H. Fukumoto, M. Onoda, H. Hayakawa, S. Ikushiro and T. Sakaki, 2011. Metabolism of the c-Fos/activator protein-1 inhibitor T-5224 by multiple human UDP-glucuronosyltransferase isoforms. Drug Metab. Dispos., 39: 803-813.
CrossRefDirect Link - Zielinska, A., C.F. Lichti, S. Bratton, N.C. Mitchell and A. Gallus-Zawada et al., 2008. Glucuronidation of monohydroxylated warfarin metabolites by human liver microsomes and human recombinant UDP-glucuronosyltransferases. J. Pharmacol. Exp. Ther., 324: 139-148.
CrossRefDirect Link - Lv, X., J. Hou, Y.L. Xia, J. Ning and G.Y. He et al., 2015. Glucuronidation of bavachinin by human tissues and expressed UGT enzymes: Identification of UGT1A1 and UGT1A8 as the major contributing enzymes. Drug Metab. Pharmacokinet., 30: 358-365.
CrossRefDirect Link - Yamanaka, H., M. Nakajima, M. Katoh and T. Yokoi, 2007. Glucuronidation of thyroxine in human liver, jejunum and kidney microsomes. Drug Metab. Dispos., 35: 1642-1648.
CrossRefDirect Link - Muzeeb, S., S.J.S. Basha, D. Shashikumar, R. Mullangi and N.R. Srinivas, 2006. Glucuronidation of DRF-6574, hydroxy metabolite of DRF-4367 (a novel COX-2 inhibitor) by pooled human liver, intestinal microsomes and recombinant human UDP-glucuronosyltransferases (UGT): Role of UGT1A1, 1A3 and 1A8. Eur. J. Drug Metab. Pharmacokinet., 31: 299-309.
CrossRefDirect Link - Tan, B., W. Cai, J. Zhang, N. Zhou and G. Ma et al., 2014. Identification of UDP-glucuronosyltransferase isoforms responsible for leonurine glucuronidation in human liver and intestinal microsomes. Xenobiotica, 44: 775-784.
CrossRefDirect Link - Shiraga, T., K. Yajima, K. Suzuki, K. Suzuki and T. Hashimoto et al., 2012. Identification of UDP-glucuronosyltransferases responsible for the glucuronidation of darexaban, an oral factor Xa inhibitor, in human liver and intestine. Drug Metab. Dispos., 40: 276-282.
CrossRefDirect Link - Liu, H.M., Z.F. Wu, Z.G. Ma and B.J. Wu, 2014. Glucuronidation of macelignan by human liver microsomes and expressed UGT enzymes: Identification of UGT1A1 and 2B7 as the main contributing enzymes. Biopharmaceut. Drug Dispos., 35: 513-524.
CrossRefDirect Link - Sun, H., H. Wang, H. Liu, X. Zhang and B. Wu, 2014. Glucuronidation of capsaicin by liver microsomes and expressed UGT enzymes: Reaction kinetics, contribution of individual enzymes and marked species differences. Expert Opin. Drug Metab. Toxicol., 10: 1325-1336.
CrossRefDirect Link - Ramirez, J., S. Mirkov, L.K. House and M.J. Ratain, 2015. Glucuronidation of OTS167 in humans is catalyzed by UDP-glucuronosyltransferases UGT1A1, UGT1A3, UGT1A8, and UGT1A10. Drug Metab. Dispos., 43: 928-935.
CrossRefDirect Link - Zhou, H., R. Shi, B. Ma, Y. Ma and C. Wang et al., 2013. CYP450 1A2 and multiple UGT1A isoforms are responsible for jatrorrhizine metabolism in human liver microsomes. Biopharm. Drug Dispos., 34: 176-185.
CrossRefDirect Link - Lee, H.S., H.Y. Ji, E.J. Park and S.Y. Kim, 2007. In vitro metabolism of eupatilin by multiple cytochrome P450 and UDP-glucuronosyltransferase enzymes. Xenobiotica, 37: 803-817.
CrossRefDirect Link - Hanioka, N., Y. Kinashi, T. Tanaka-Kagawa, T. Isobe and H. Jinno, 2017. Glucuronidation of mono (2-ethylhexyl) phthalate in humans: Roles of hepatic and intestinal UDP-glucuronosyltransferases. Arch. Toxicol., 91: 689-698.
CrossRefDirect Link - Kemp, D.C., P.W. Fan and J.C. Stevens, 2002. Characterization of raloxifene glucuronidation in vitro: Contribution of intestinal metabolism to presystemic clearance. Drug Metab. Dispos., 30: 694-700.
PubMedDirect Link - Lu, S.H., J.W. Wu, H.L. Liu, J.H. Zhao and K.T. Liu et al., 2011. The discovery of potential acetylcholinesterase inhibitors: A combination of pharmacophore modeling, virtual screening and molecular docking studies. J. Biomed. Sci., Vol. 18.
CrossRefDirect Link - Ako, R., D. Dong and B. Wu, 2012. 3D-QSAR studies on UDP-glucuronosyltransferase 2B7 substrates using the pharmacophore and VolSurf approaches. Xenobiotica, 42: 891-900.
CrossRefDirect Link - Sakkiah, S., A. Baek and K.W. Lee, 2012. Pharmacophore modeling and molecular dynamics simulation to identify the critical chemical features against human sirtuin 2 inhibitors. J. Mol. Struct., 1011: 66-75.
CrossRefDirect Link - Ekins, S., G. Bravi, S. Binkley, J.S. Gillespie, B.J. Ring, J.H. Wikel and S.A. Wrighton, 2000. Three- and Four-Dimensional-Quantitative Structure Activity Relationship (3D/4D-QSAR) analyses of CYP2C9 inhibitors. Drug Metab. Dispos., 28: 994-1002.
Direct Link - Smith, P.A., M.J. Sorich, R.A. McKinnon and J.O. Miners, 2003. Pharmacophore and quantitative structure-activity relationship modeling: Complementary approaches for the rationalization and prediction of UDP-glucuronosyltransferase 1A4 substrate selectivity. J. Med. Chem., 46: 1617-1626.
CrossRefDirect Link - Kola, I. and J. Landis, 2004. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov., 3: 711-716.
CrossRefDirect Link - Roberts, S.A., 2001. High-throughput screening approaches for investigating drug metabolism and pharmacokinetics. Xenobiotica, 31: 557-589.
CrossRefDirect Link