
Mohammed Hussen Bule
Department of Pharmacy, College of Medicine and Health Sciences, Ambo University, Ambo, Ethiopia
Ishtiaq Ahmed
Department of Biochemistry, University of Agriculture Faisalabad, Pakistan
Faheem Maqbool
School of Pharmacy, Pharmacy Australia Centre of Excellence, University of Queensland, Brisbane, QLD 4102, Australia
Muhammad Anjum Zia
Department of Pharmacy, College of Medicine and Health Sciences, Ambo University, Ambo, Ethiopia
International Journal of Pharmacology
Year: 2017 | Volume: 13 | Issue: 7 | Page No.: 818-831







ABSTRACT
Malaria causes over a million deaths each year (2 percent of the global total of deaths), with hundreds of millions of clinical episodes per annum. The greatest challenge to malaria control and eradication is the emergence of malaria parasites that are resistant to antimalarial drugs. The development of resistance to conventionally used anti-malarial drugs, such as chloroquine (CQ) and Sulfadoxine-Pyrimethamine (SP) has been documented. To counter this WHO recommended that artemisinin-based combination therapy (ACT) should be used for treating uncomplicated Plasmodium falciparum malaria to ensure efficacy and reduce the emergence of drug-resistant parasites. Currently available antimalarial drugs are ineffective and their number is declining because of the widespread resistance. Thus, the new antimalarial agent is in urgent demand; however, the development of new antimalarial drug presents challenges due to resistance, toxicity, minimal efficacy of those on the pipeline and high cost of drug research. Identification of novel drug targets and design of new chemical compounds acting on new targets is important to control the emergence of resistance to existing drugs. In this regard, a natural product derived synthetic analogs of febrifugine containing quinazolinone scaffold can be considered best. Therefore, quinazolinones are potential compounds in seeking for novel drugs that act against the malarial pathogen. Hence, in this review compounds containing quinazolinone structure and possessing antimalarial activities are covered.
PDF Abstract XML References Citation
How to cite this article
Mohammed Hussen Bule, Ishtiaq Ahmed, Faheem Maqbool and Muhammad Anjum Zia, 2017. Quinazolinone Derivatives as a Potential Class of Compounds in Malaria Drug Discovery. International Journal of Pharmacology, 13: 818-831.
DOI: 10.3923/ijp.2017.818.831
URL: https://scialert.net/abstract/?doi=ijp.2017.818.831
DOI: 10.3923/ijp.2017.818.831
URL: https://scialert.net/abstract/?doi=ijp.2017.818.831
INTRODUCTION
Malarial symptoms are described in ancient Chinese and Sanskrit medical texts and Hippocrates referred to the disease in the 4th Century BC1. It was associated with ‘bad air’ by the 18th century Italians meaning malaria from where the name malaria is derived2,3. In children under age five malaria accounts for almost 1 in 10 deaths worldwide and 1 in 5 deaths in Sub-Saharan Africa (SSA), which makes it one among the leading killers4. More than 80% of deaths due to malaria occurs in SSA where 90% accounts for children under five5. Malaria causes over a million deaths each year (2 percent of the global total of deaths), with hundreds of millions of clinical episodes per annum3. Malaria also places a tremendous burden on national health systems and individual families6. In the governmental budget proportions allocated to health system ranges from 5% in Africa, Asia and the Eastern Mediterranean Region, to well over 20% in some countries in Americans7. The economic impact of malaria is disproportionately felt by the poor. A study in Tanzania indicated that the death due to acute fever among children of the poorest families was 39% higher than among the wealthiest children3. The Plasmodium parasite, responsible for causing malaria, is a protozoan with four identified species which causes human malaria via a female Anopheles mosquito as a vector, namely: P. falciparum, P. vivax, P. malariae and P. ovale2. Even though these four species of plasmodium parasite can infect human and cause malaria, P. falciparum is the riskiest and potentially threatening8. However, Plasmodium vivax also creates significant human morbidity, suffering and economic loss, being responsible for 70 million to 80 million cases of the global malaria burden each year9.
Epidemiology of malaria: There are 109 malaria endemic countries in tropical and subtropical zones, across all countries except Antarctica and Australia. In these countries, the intensities of transmission vary from very low to extremely high10. Human malaria infections caused an estimated 214 million clinical cases and 438,000 deaths in 2015. The relatively low case-fatality rate, even for the most virulent species, P. falciparum, is partly due to patient immunity acquired after repeated infections but is also attributable to the timely provision of effective malaria drugs11. The SSA region is the hardest hit by malaria comprising endemic areas of stable transmission12. The millennium development goal Target 8 "Global partnership for development" ("Have halted by 2015 and begin to reverse the incidence of malaria and other major diseases [including TB]") were to be met before 201513. Whereas, the target set under Millennium Development Goal 6 was to be reached by 55 countries that are on track to reduce their malaria burden by 75%. Despite progress in the reduction of malaria morbidity and mortality in recent years, malaria remains one of the leading health problems in endemic countries14. Global malaria death estimates in the 1980s and 1990s range from 800 000 to almost 2.5 million; in the 2000s, the range is from 650 000 to more than 1 million. Studies showed that malaria is the underlying cause of death for 1.24 million individuals, including 714 000 children younger than 5 years and 524 000 individuals aged 5 years or older in201015. Children in sub-Saharan Africa and Southeast Asia have the highest risk of contracting and dying from malaria16. Being a curable disease, early diagnosis and prompt treatment is a key strategy to reduce morbidity and mortality from malaria. A core component of any malaria elimination program is to ensure that all patients with malaria are rapidly diagnosed, have access to highly effective antimalarial drugs14. Much of this morbidity and mortality could be avoided if drugs available to patients were efficacious, high quality and used correctly16. During the past 5 years, substantial progress has been made in the fight against malaria, with a 31% reduction in global malaria deaths15.
Life cycle of malaria: Malaria parasites live in mosquito and human hosts and go through several developmental and transmission phases on the way to causing disease in humans 17. Malaria is a mosquito-borne infectious disease transmitted by a parasitic protozoan in the Plasmodium genus18. The malaria parasite is a single-cell protozoan (plasmodium). Members of the genus Plasmodium have a complex life cycle, Fig. 1. A sexual stage occurs within the Anopheles mosquito, while asexual stages take place in the host. Malaria is transmitted from one human to another through the insect vector, the female Anopheles mosquito19,20. Only the female mosquito bites because it needs blood to produce eggs. Mosquitoes bite a variety of hosts-birds, dogs, horses, cattle and people18. The infection of human erythrocytes is ultimately responsible for all the clinical pathologies associated with the disease17.
Initially, when female Anopheles mosquito bites an infected human, it gets infected and intakes gametocytes. The sexual transformation of gametocytes into ookinetes and ookinetes into oocyst takes place inside the midgut of mosquito.
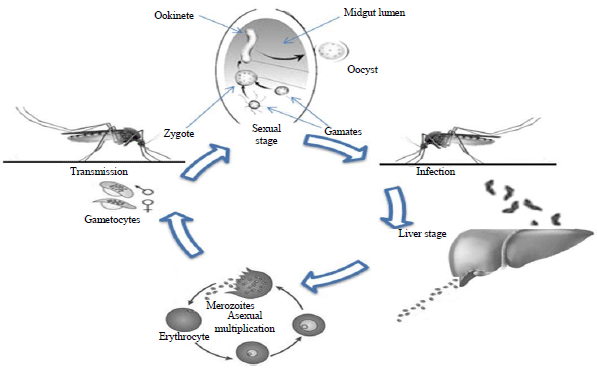 | |
| Fig. 1: | Life cycle of the malaria parasite |
Finally, sporozoites are developed from oocysts, which eventually burst, releasing sporozoites into the salivary gland21. At the mosquito’s next feeding, the sporozoites are injected into the blood stream of another human to begin the asexual stages. Once the mosquito inoculates the parasites (sporozoites) into the bloodstream, the parasites invade the liver within 30 min and start replicating there (schizonts). Also, P. vivax and P. ovale can remain dormant in the liver (hypnozoites, not shown in Fig. 1) and cause relapses years after the initial infection22. After a relatively brief residence (less than an hour) in the systemic circulation, the sporozoites invade liver parenchymal cells, where they divide and develop asexually into multinucleated schizonts. These are the primary exoerythrocytic tissue forms of the parasite. When this primary stage of development is completed (6-2 days), the schizonts will rupture, releasing merozoites into the blood. These latter forms invade host erythrocytes, where they again grow and divide asexually (erythrocytic schizogony) and become red cell schizonts. Some of the parasites differentiate into sexual (male and female) forms or gametocytes. If the diseased human is bitten by a mosquito at this time, the gametes will be taken up into the organism’s gut to repeat the reproductive cycle19.
Prevention and control of malaria: Since the launch of the Roll Back Malaria initiative by WHO in 1998 and particularly in the past few years, malaria control has intensified in endemic countries, supported by a greatly increased investment of financial resources and technical assistance from the international community10. Three main strategies are presently attempting to control the disease: Vaccination, vector control and parasitical drugs. Of these, parasitical drugs are currently the main line of disease control until vaccination or mosquito control can be implemented more successfully23. To eradicate malaria, achievable milestones must be set. The publication of the Global Malaria Action Plan (GMAP) by the WHO set some of those milestones; for example, a tenfold reduction in malaria incidence and deaths by 2030 (compared with 2015). The first GMAP was published in 2008 and covered the period until 2015. More recently, input and consultations are being sought from experts and regions for GMAP2. Both GMAP2 and the Global Technical Strategy for Malaria (coordinated by the WHO) will cover the 10 years between 2016 and 202524. On Oct 17, 2007, Bill and Melinda Gates called for complete eradication to be adopted as the new goal for the age-old fight against malaria, with the Director-General of WHO, Margaret Chan, promptly echoing their conviction. Two crucial questions stand out for those organizations that will now begin striving towards malaria eradication. When and how can it be achieved?24,25.
During the past decade, a range of organizations has led a global movement to combat malaria. Accurate assessments of the levels and time trends in malaria burden are crucial for the evaluation of progress towards goals and the focusing of future efforts15. A report released by WHO finds that the global burden of malaria remains enormous but that access to malaria control interventions, especially bed nets in Africa, increased sharply between 2004 and 200626.
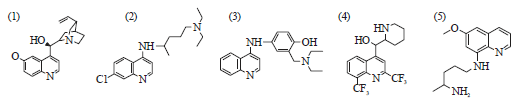 | |
| Fig. 2: | Chemical structures of some quinoline derivatives |
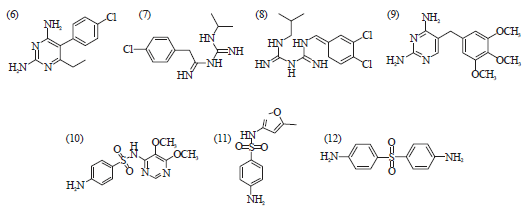 | |
| Fig. 3: | Chemical structures of antifolate drugs |
As a result of malaria control efforts across the world, 80 countries are now in the phase of malaria control; 12 countries are making the program transition to elimination; 11 countries are operating malaria elimination programs and 6 countries are actively engaged in preventing re-introduction of malaria10. Despite extensive control efforts, the incidence of the disease is not decreasing in most malaria-endemic areas of the world and some it is increasing27.
Drugs for treatment of malaria: Currently available antimalarial agents comprise classes of drugs classified based on their chemical structures and mechanism of action28. The number of antimalarial drugs in use today is limited due to the wide spread of resistance. Those in use to date are the quinine derivatives, the artemisinins and antifolate combination drugs.
Quinoline derivatives: The quinoline derivatives have long been used for the treatment of malaria, the first one being quinine which is isolated from the bark extract of Cinchona trees. In the 17th century, the pulverized bark was widely used in Europe. As the quest for new compounds for treating malaria was on the rise the first 4(8), -aminoquinolines derivatives were developed29. The success of the antimalarial aminoquinoline drugs has been based on excellent clinical efficacy, limited host toxicity, ease of use and simple, cost-effective synthesis 30. The 4-aminoquinoline derivatives of quinine (1), shown in Fig. 2 are chloroquine (2), amodiaquine (3) and mefloquine (4), whereas primaquine (5) is an 8-aminoquinoline derivative of quinine31. The mode of action and mechanism of resistance of these derivatives is not fully understood since much of the focus is directed to the identification of novel chemotherapeutic agents32. The quinolones are known to inhibit the polymerization of heme and prevent disposal of polymers from the food vacuole to the cytoplasm where hemozoin is formed. This leads to antiparasitic accumulation of free heme, which is highly toxic to the parasite33.
Antifolate combination drugs: Antifolates are various combinations of dihydrofolate reductase inhibitors (proguanil (6), pyrimethamine (7), chlorproguanil (8) and trimethoprim (9)) and sulfa drugs (sulfadoxine (10), sulfamethoxazole (11), dapsone (12) and others), Fig. 3. Most commonly used combinations include sulfadoxine-pyrimethamin and sulfamethoxazole-trimethoprim. Currently, an antifolate combination drug, dapsone and chlorproguanil was tested and has a more potent synergistic antimalarial action than other drugs such as sulfadoxine-pyrimethamine31.
Antibiotics: Tetracyclines (13 and 14) Fig. 4, usually in conjunction with quinine (1), have been reported to be suppressive in human malaria and proved to be of value as an additional drug for the radical cure of chloroquine-resistant falciparum infections.
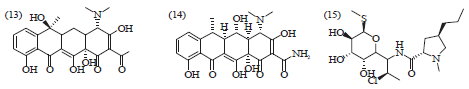 | |
| Fig. 4: | Chemical structures of antimalarial antibiotics |
 | |
| Fig. 5: | Chemical structures of artemisinin and related compounds |
 | |
| Fig. 6: | Chemical structures of napthoquinone, phenanthrene and fluoromethanol derivatives |
Clindamycin (15), alone or in combination with quinine, has also been used for the treatment of CQR falciparum malaria in Thailand with good results, despite the side effects. In non-immune individuals, its efficacy has not been fully established 34.
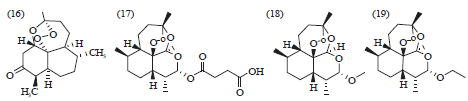
Artemisinin compounds: Artemisinin (qinghaosu) (16), artesunate (17), artemether (18) and arteether (19) have all been used alone or in combination therapy as antimalarial agents35. Artemisinin (16) Fig. 5, which is present in the extracts of the aerial parts of the plant Artemissia annua, has been utilized for more than a millennia for fever and rediscovered as antimalarial agent36. The emergence of multidrug-resistant strains of P. falciparum and chloroquine-resistant strains of P. vivax in malaria endemic areas emphasize on preparing new, efficient and affordable antimalarial medications. Thus, Artemisinin Combination Therapy (ACT) was recommended by WHO for successful treatment of uncomplicated malaria37.
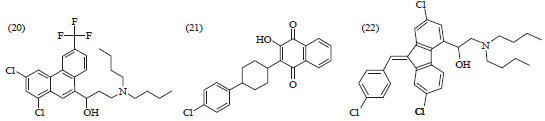
Other compounds with antimalarial activity: Halofantrine (20), which consists phenanthrene-methanol and illustrated in Fig. 6, is active against the erythrocytic stage of malaria. In areas of multi-drug resistant falciparum malaria, halofantrine is mainly recommended. Atovaquone (21), which is a hydroxynaphthoquinone, is most widely used for treating opportunistic infections in immune-compromised patients. It is also effective against CQR P. falciparum. However, it is usually given in combination with proguanil to avoid the development of resistance. Lumefantrine (22), a fluoro methanol compound, is being produced as a fixed combination tablet with artemether [25]31.
Antimalarial drug resistance: Malaria continues to pose a challenge given its resurgence and problem of drug resistance. Chloroquine resistance in P. falciparum was first detected in Thailand in 1962 and India in 197338. Since 2000, malaria-associated mortality has been reduced by more than 50% but emerging drug- and insecticide-resistance continues to pose a major threat24. The development drug resistance has jeopardized the efficacy of every antimalarial drug in use today. In fact, the history of malarial drug developments parallels the history of drug resistance development39. The WHO defined antimalarial drug resistance as the "ability of a parasite strain to survive and multiply despite the administration and absorption of a drug given in doses equal to or higher than those usually recommended but within a tolerance of the subject21”. Broadly, there are two ways in which malaria parasites have become resistant to antimalarial drugs. Resistance against antifolates and atovaquone has arisen by mutations in drug targets that reduce their sensitivity; in these examples, an understanding of the molecular basis of drug action has been a prerequisite for elucidating the mechanism of drug resistance. Other drugs such as chloroquine and mefloquine may not have parasite-derived protein targets that can mutate, allowing parasites to escape from therapies. For these classes of antimalarial drugs, the parasite has become resistant through mutations in transporters involved in determining drug disposition within the intraerythrocytic parasite and its organelles. This effectively reduces drug concentrations at critical (presumed) target sites40.
The development of resistance to conventionally used anti-malarial drugs, such as chloroquine (CQ) and Sulfadoxine-Pyrimethamine (SP) has been documented14. Chloroquine strength and high-level sulfadoxine-pyrimethamine resistance in P. falciparum both originated in South-East Asia and subsequently spread to Africa37. Currently, scientists reported that they had found the first evidence of resistance to the world's most efficient drug coartem (lumefantrine and artemether) for treating malaria in western Cambodia31. Consequently, another change to artemether-lumefantrine was suggested in 200441. WHO recommended that artemisinin-based combination therapy (ACT) should be used for treating uncomplicated Plasmodium falciparum malaria to ensure efficacy and reduce the emergence of drug-resistant parasites14,24. Several ACT combinations available include artemether-lumefantrine (AL), artesunate-amodiaquine (AS/AQ), artesunate-mefloquine (AS/MQ), artesunate-chlorproguanil-dapsone (AS/CD), artesunate-sulphadoxine-pyrimethamine (AS/SP), dihydroarte misinin-piperaquine (DA/PQ), artesunatepiperazine (AS/PZ) and artesunate-atovaquone-proguanil (A/AP). Out of these ACT combinations WHO recommended AL, AS/MQ, AS/AQ and AS/SP. Several countries have now adopted ACTs as the first line agents for uncomplicated malaria42.
The concept of combination therapy relies on the rapid onset of schizonticidal action to rapidly reduce parasitaemia, leaving the residual parasitaemia to be cleared by high concentrations of the partner drug14. However, there is a constant threat of malaria evolving resistance to available drugs and recent observations that resistance may have arisen to the most widely used antimalarial drug class, the artemisinins11. Although ACTs are designed to reduce the chance of artemisinin drug resistance development, there are considerable concerns that this may already have occurred. For instance, there is now mounting evidence that the efficacy of artemisinin derivatives is reduced in Southeast Asia, where artemisinin derivatives have been used for a long time as monotherapies43.
The greatest challenge to malaria control and eradication is the emergence of malaria parasites that are resistant to antimalarial drugs21. The major drug resistance problem occurs with P. falciparum, which is of particular concern because of the enormous burden of disease caused by this species, its lethal potential, the propensity for epidemics and the cost of candidate replacement drugs for areas with established drug resistance37. Whereas, other less lethal strains, P. ovale and P. vivax, can exist as latent hypnozoites in the liver which can initiate a relapse months to years after the initial infection44. Plasmodium falciparum can be clinically resistant to all monotherapy with current antimalarial drugs. In South-East Asia, the combination of quinine and tetracycline is the treatment of choice for multidrug-resistant P. falciparum infections45.
Future drug candidates that are developed with the aim to circumvent or stall resistance will help produce the next generation of therapies that prevent or reduce mortality39. Any new antimalarial drug that is developed will ultimately be delivered as a combination therapy to delay the likely emergence of parasite resistance. Compounds in combinations should ideally act against different cellular targets to offset the likelihood that a single parasite genome-amplification event could render parasites resistant to both drugs46. Furthermore, poor-quality antimalarial drugs are very likely to jeopardize the unprecedented progress and investments in control and elimination of malaria made in the past decade. Of the many public health consequences of poor-quality antimalarial drugs, drug resistance is a particular concern. Low concentrations of active pharmaceutical ingredient in a bad quality antimalarial drugs can result in sub-therapeutic concentrations of drug in vivo, which contributes to the selection of resistant parasites 16. Thus, we need to protect the medicines we have by ensuring correct deployment and continual vigilance to stop the production and distribution of counterfeit medicines. Even with fixed-dose combinations, there remains a risk of resistance emerging and therefore a need for new medicines. New molecules should shorten the duration of treatment and increase compliance and also prevent the transmission of the parasite back to the insect vector24. Therefore, there is an urgent need to gain information about the basic mechanisms through which antimalarial drugs act and resistance is generated in order not only to identify new targets and develop new drugs with novel mechanisms of action but also to take advantage of the mode of action of available drugs and make better use of them47.
Recent advances in antimalarial drug development: Mefloquine is the only synthetic antimalarial agent discovered over the past 30 years.
 | |
| Fig. 7: | Chemical structures of fosmidomycin, chalcone, naphthoquinone and arylsulfonyl acridinyl derivatives |
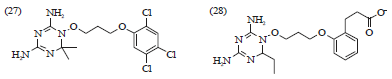 | |
| Fig. 8: | DHFR inhibitors |
 | |
| Fig. 9: | 4-aminoquinoline antimalarials |
Whereas, artimisisin, which is discovered in this period is a natural product, whose medicinal actions have been known for over 2 millennia48. However, the sudden resurgence of malaria and emergence of malarial drug resistance in many countries of the world have made the synthetic efforts toward new antimalarial drugs very important28. Recently some synthetic compounds are reported to have potent antimalarial activity against different Plasmodium species.
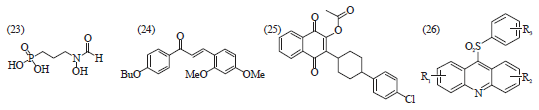
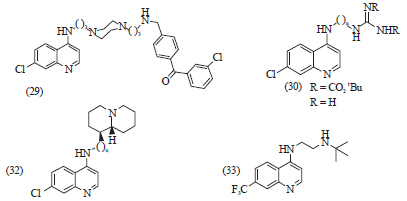
Clinical trials conducted with fosmidomycin (23) in combination with clindamycin (15) or artesunate (17) have shown high efficiency in the treatment of acute, uncomplicated malaria48. Recently synthetic chalcone analog, 2,4-dimethoxy-4 butoxychalcone (24), was reported to have excellent antimalarial activity49,50. In addition to this, the antimalarial activity of naphthoquinone derivatives (25), Fig. 7, has been widely reported. Acylation of the hydroxy moiety of atovaquone (21) led to a compound exhibiting similar activity as atovaquone (21) and more active than chloroquine (2) and quinine (1) against P. falciparum51. Arylsulfonyl acridinyl derivatives (26), having an acridinic ring and aryl sulfone moiety together are also reported to have an antimalarial activity on P. falciparum52. Structure-based drug design resulted in P218 (28), a DHFR inhibitor active against all clinically relevant mutations. P218 (28) combines the pyrimidine ring of pyrimethamine (Fig. 8), which brings potency and the linker of the DHFR inhibitor WR99210 (27), which tolerates mutations due to its flexibility. The P218 is more potent than pyrimethamine against DHFR in the wild-type strain TM4 (IC50 = 4.6 and 58 nM, respectively) as well as in the quadruple mutant strain V1/S (IC50 = 56 and >100,000 nM, respectively)22. Recently, a series of bisquinolines (29, 30, 31, 32 and 33, Fig. 9), where the 4-aminoquinoline part of chloroquine was retained and bisamide links joined the two units, have been synthesized and screened against CQS and CQR strains of P. falciparum in vitro. The resistant indices for all the compounds were found to be lower than that of CQ. The position of attachment and length of the linker chain had marked effect on the activity33.
Plasmepsins: Potential drug targets for antimalarial drugs: With the technological developments of the past few decades, the ability to search for new drug candidates has rapidly accelerated. Advances in robotic automation and liquid handling, coupled with the ever-shrinking scale at which these assays are performed, have facilitated ultra-HTS of enormous compound libraries46.
 | |
| Fig. 10: | Plasmepsin inhibitors |
Identification of novel drug targets and design of new chemical compounds acting on new targets is nowadays widely used approach all over the world to combat issue raised by the emergence of resistance to existing drugs. Therefore, investigating inhibitors specific for the new target proteins of malaria parasite has been exploited for drug target identification and currently studies are underway53. Thus far, the genetics underlying the new resistance against artemisinins are becoming increasingly understood and this knowledge is being used to set up panels of parasites against which new drug candidates can be tested54. Detecting the different genetic basis for malaria not only reveals the disease pathogenesis but also facilitates discovering new targets for anti-malaria drugs55. There is an argument to be made that hitting the sexual stages is useful- this blocks transmission and may delay resistance since there are several orders of magnitude fewer parasites from which to select mutants56. Given that the liver stage would be highly desirable for candidate drugs to have activity against hepatic and sexual forms of the malarial parasite, it is surprising that few clinical trials, to date, have examined whether gametocyte carriage can be reduced following drug treatment57.
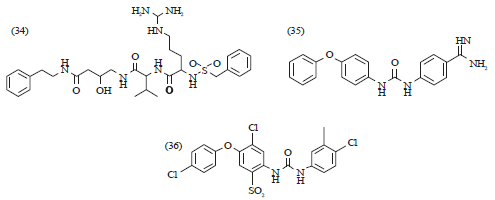
Because proteases play important roles during parasite infection of and development in the mosquito, they were considered as potential transmission-blocking targets. Transcriptomic data suggested that Plasmodium aspartic proteases, known as plasmepsins are expressed in sexual stage parasites58. There are 10 plasmepsins in P. falciparum. Other Plasmodium species have only seven; they have only one digestive vacuole plasmepsin instead of the four in falciparum56. Plms I-IV are the most studied isoforms owing to their expression and important role during the blood stage. Whereas Plm II remains the best-studied isozyme, Plms V-X remain considerably less understood; recent studies show that Plm V functions as Plasmodium export element (PEXEL)-cleaving protease for protein export from the food vacuole to the erythrocyte to enable the development of P. falciparum parasites59,60. Expression of Plm I, II, IV, V, IX, X and HAP occurs in the erythrocytic stage, whereas Plm VI, VII and VIII are expressed in the exo-erythrocytic stages. The digestive vacuole plasmepsins are 55-75% identical to each other but 10-25% identical to the other plasmepsins61. It has been shown that the general aspartic proteinase inhibitor isovaleryl pepstatin is a tight binding (sub-nanomolar Ki) inhibitor of plasmepsin I and plasmepsin II. However, this compound inhibits most aspartic proteinases and so has no value as a potential drug62. Plasmepsin V is an aspartic acid protease expressed by protozoan parasites of Plasmodium species and it has a crucial role in recognizing and processing effector proteins for export to host cells. The most potent plasmepsin V inhibitor to date, WEHI-916 (34), has a high affinity for the endogenous enzyme but has a modest ability to inhibit P. falciparum growth63. Other small nonpeptide inhibitors of Plm II based on a diphenylurea unit were discovered by a screening of compounds in the Walter Reed chemical database. BothP. Falciparum Plm II and P. Vivax plasmepsin were used in the screening assays. Compounds 35 and 36, Fig. 10, were identified as the lead inhibitors with great high-plasmepsin potency61.
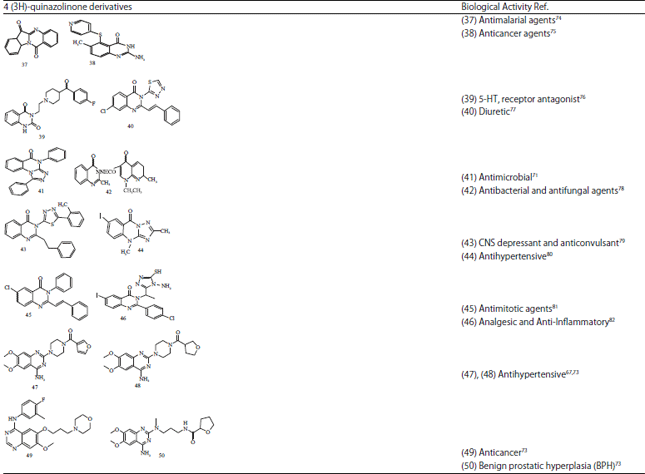
Review of quinazolinones: Various structural class of compounds has been reported to possess antimalarial activities such as chalcones51, thienopyrimidinone64, quinolones, quinazolins47, enaminones65, acridines48. Among the different heterocyclic structures which have been studied for their antiplasmodial properties, quinazoline has quite recently shown increasing interest66. Quinazolinones are versatile nitrogen heterocyclic compounds, displaying wide applications including anticonvulsant, sedative, tranquilizer, analgesic, antimicrobial67, anesthetic, antioxidant68, anticancer, antiviral67, anti-TB69, antihypertensive, anti-inflammatory, antimalarial, diuretic and muscle relaxant properties, Table 1.
| Table 1: | Some 4 (3H)-quinazolinone derivatives and their biological activity |
 | |
Increased efforts in antimalarial drug discovery are urgent to develop safe and affordable new drugs to counter the spread of malaria parasites that are resistant to existing agents. Furthermore, quinazolinones substituted at 2 and 3-position plays a pivotal role in the antimalarial activity70-72. Moreover, there are marketed quinazoline derivatives such as prazocin and trazocin as antihypertensive agents67,73 and Gentifib as anticancer73 and alfuzosin for treating benign prostatic hyperplasia, which gives hope to research in classes of compounds73.
Febrifugines derivatives and other important quinazolinones: The isomeric alkaloids (+)-febrifugine (51) and (+)-isofebrifugine (52), Figure 11 are found in the roots and leaves of the Chinese medicinal plant Dichroa febrifuga (also called Chinese quinine) belonging to the Saxifragaceae family83. Several bio-active natural products such as febrifugine (51) and isofebrifugine (52) contain quinazolinone moieties with potential antimalarial activity83. Compound 53 with an extra nitrogen atom in the position 5 or 6 of the aromatic ring (IC50= 1.2 nM) possessed antimalarial activity comparable to 51, while compound 54 (IC50 = 0.33 nM) with difluoride attached to C-5 and C-6 was superior to febrifugine. These compounds were 100 times less toxic than febrifugine84. In one study it was reported that antimalarial activity of a series of febrifugine derivatives bearing structural modifications at (i) the quinazoline ring, (ii) the linker or (iii) the piperidine ring. Thienopyrimidine analog 55 exhibited potent antimalarial activity and a high therapeutic selectivity both in vitro and in vivo [EC50 = 0.00306 lg mL–1 (P. falciparum FCR-3), ED50 = 2.95 mg kg–1, LD50 = 88 mg kg–1 (P. berghei)]85. The fluorinated analog 57, which was designed to prevent metabolic oxidation, actually showed higher antimalarial activity than that of (+)-febrifugine (51) but it also proved more toxic. On the other hand, analog 55 showed high in vitro and in vivo antimalarial activity.
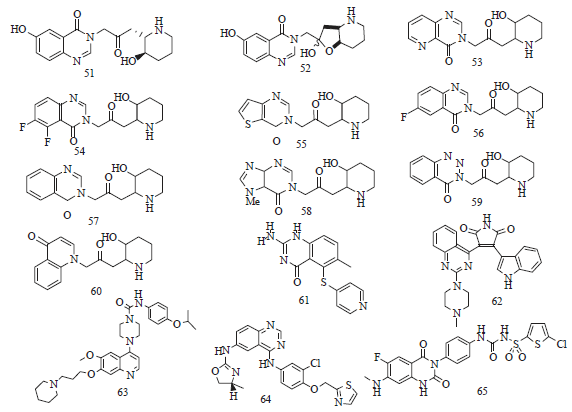 | |
| Fig. 11: | Quinazolines, Quinazolinones, and analogs of febrifugine |
However, the other analogs, 57-59, showed little antimalarial activity with total loss of antimalarial activity being observed for 60. The complete loss of antimalarial activity in analog 60 indicates that a basic nitrogen group within the heteroaromatic portion is essential for antimalarial activity85-87. Febrifugine acts by impairing hemozoin formation required for maturation of the parasite at the trophozoite stage. The use of febrifugine as an anti-malarial agent is initially appealing not only because of its rapid effect and no drug resistance but also because of its availability. Subsequent pre-clinical researchers have found that febrifugine possesses adverse side effects. Strong liver toxicity has precluded febrifugine as a clinical drug22,69,88. There are some available marketed drugs possessing quinazoline and quinazolinone ring such as the anti-solid tumor Nolatrexed (61), Sotrastaurin (62) used for psoriasis and ulcerative colitis, Tandutinib (63) for gloiblastoma, Varlitinib (64) for anticancer and Elinogrel (65) as antithrombosis70.
CONCLUSION
Globally the importance fighting malaria is recognized. However, the burden of malaria is still high in developing countries, especially in the Sub-Saharan African region. Although decades-long efforts and academic engagements have been there, malaria continues to pose a challenge given its resurgence and problem of drug resistance. Most of the existing antimalarial drugs including chloroquine are brought to the sideline by the emergence of resistance. Currently, a combination therapy is recommended by WHO to reduce the risk of drug resistance. However, there are still reports indicating drug resistance even to those used in combination recommended by WHO such as artimisinin. Thus, malaria drug discovery is unquestionably urgent to battle against the disease. To minimize the likely hood of cross-resistance the focus of the drug discovery must be on newer drug targets, such as plasmepsin. In finding a novel antimalarial agent, a various library of chemicals has been investigated and heterocyclic compounds such as chalcones, thienopyrimidinone, quinolones, quinazolines, quinazolinones, enaminones and acridines have been reported to possess antimalarial activity. Further, assessment on quinazolinones has shown fruitful results and their activity as an antimalarial agent is appealing. In this regard, febrifugine and its derivatives have been reported to have excellent activity. Therefore, quinazolinones can be considered potential compounds in seeking for novel drugs that act against the malarial pathogen.
SIGNIFICANCE STATEMENTS
Quinazolinone derivatives possess broad spectrum activities including antimalarial, antibacterial, antifungal, antiviral, anti-HIV, anti-inflammatory and many others. Owing to their inherent bioactive functionalities, quinazolinone derivatives have been extensively exploited in various biomedical sectors for different purposes with a particular reference to malaria drug discovery. With the advent of recent technologies, quinazolinone-based pharmaceuticals can be synthesized more efficiently through using various processing approaches. This review mainly focuses on quinazolinone-based bioactive compounds and their antimalarial potentialities.
ACKNOWLEDGMENTS
All authors thank and acknowledge their respective Universities and Institutes.
REFERENCES
- De Oliveira, R.B., E.M. de Souza-Fagundes, R.P.P. Soares, A.A. Andrade, A.U. Krettli and C.L. Zani, 2008. Synthesis and antimalarial activity of semicarbazone and thiosemicarbazone derivatives. Eur. J. Med. Chem., 43: 1983-1988.
CrossRefDirect Link - Kalra, B.S., S. Chawla, P. Gupta and N. Valecha, 2006. Screening of antimalarial drugs: An overview. Indian J. Pharmacol., 38: 5-12.
CrossRefDirect Link - Bule, M.H., A. Haymete and B. Kefale, 2015. Synthesis and in-vivo pharmacological evaluation of some novel 4(3H)-quinazolinone derivatives as potential anti-malarial agents. Drug Des., Vol. 4.
CrossRefDirect Link - Diagana, T.T., 2015. Supporting malaria elimination with 21st century antimalarial agent drug discovery. Drug Discov. Today, 20: 1265-1267.
CrossRefDirect Link - Dikasso, D., E. Makonnen, A. Debella, D. Abebe and K. Urga et al., 2006. In vivo anti-malarial activity of hydroalcoholic extracts from Asparagus africanus lam. in mice infected with Plasmodium berghei. Ethiop. J. Health Dev., 20: 112-118.
Direct Link - Mbatchi, S.F., B. Mbatchi, J.T. Banzouzi, T. Bansimba and G.F.N. Ntandou et al., 2006. In vitro antiplasmodial activity of 18 plants used in Congo Brazzaville traditional medicine. J. Ethnopharmacol., 104: 168-174.
CrossRefPubMedDirect Link - Hastings, I.M., E.M. Hodel and K. Kay, 2016. Quantifying the pharmacology of antimalarial drug combination therapy. Sci. Rep., Vol. 6.
CrossRefDirect Link - Naing, C., M.A. Whittaker, J.W. Mak and K. Aung, 2015. A systematic review of the efficacy of a single dose artemisinin-naphthoquine in treating uncomplicated malaria. Malar. J., Vol. 14.
CrossRefDirect Link - Murray, C.J.L., L.C. Rosenfeld, S.S. Lim, K.G. Andrews and K.J. Foreman et al., 2012. Global malaria mortality between 1980 and 2010: A systematic analysis. Lancet, 379: 413-431.
CrossRefDirect Link - Nayyar, G.M.L., J.G. Breman, P.N. Newton and J. Herrington, 2012. Poor-quality antimalarial drugs in southeast Asia and sub-Saharan Africa. Lancet Infect. Dis., 12: 488-496.
CrossRefDirect Link - Haldar, K., S.C. Murphy, D.A. Milner and T.E. Taylor, 2007. Malaria: Mechanisms of erythrocytic infection and pathological correlates of severe disease. Annu. Rev. Pathol.: Mech. Dis., 2: 217-249.
CrossRefPubMedDirect Link - Muanda, F.T., S. Chaabane, T. Boukhris, F. Santos and O. Sheehy et al., 2015. Antimalarial drugs for preventing malaria during pregnancy and the risk of low birth weight: A systematic review and meta-analysis of randomized and quasi-randomized trials. BMC Med., Vol. 13.
CrossRefDirect Link - Biamonte, M.A., J. Wanner and K.G. Le Roch, 2013. Recent advances in malaria drug discovery. Bioorg Med. Chem. Lett., 23: 2829-2843.
CrossRefPubMedDirect Link - Ralph, S.A., M.C. D'Ombrain and G.I. McFadden, 2001. The apicoplast as an antimalarial drug target. Drug Resistance Updates, 4: 145-151.
CrossRefDirect Link - Wells, T.N.C., R.H. van Huijsduijnen and W.C. Van Voorhis, 2015. Malaria medicines: A glass half full? Nat. Rev. Drug Discov., 14: 424-442.
CrossRefDirect Link - Feachem, R. and O. Sabot, 2008. A new global malaria eradication strategy. The Lancet., 371: 1633-1635.
CrossRefDirect Link - Ballou, W.R., M. Arevalo-Herrera, D. Carucci, T.L. Richie and G. Corradinet al., 2004. Update on the clinical development of candidate malaria vaccines. Am. J. Trop. Med. Hyg., 71: 239-247.
PubMedDirect Link - Rosenthal, P.J., 1998. Proteases of malaria parasites: New targets for chemotherapy. Emerg. Infect. Dis., 4: 49-57.
PubMedDirect Link - Vangapandu, S., M. Jain, K. Kaur, P. Patil, S.R. Patel and R. Jain, 2007. Recent advances in antimalarial drug development. Med. Res. Rev., 27: 65-107.
CrossRefDirect Link - Delarue-Cochin, S., E. Paunescu, L. Maes, E. Mouray, C. Sergheraert, P. Grellier and P. Melnyk, 2008. Synthesis and antimalarial activity of new analogues of amodiaquine. Eur. J. Med. Chem., 43: 252-260.
CrossRefDirect Link - Smith, R.D. and J. Coast, 2002. Antimicrobial resistance: A global response. Bull. World Health Organiz., 80: 126-133.
Direct Link - Kaur, K., M. Jain, R.P. Reddy and R. Jain, 2010. Quinolines and structurally related heterocycles as antimalarials. Eur. J. Med. Chem., 45: 3245-3264.
CrossRefDirect Link - Wyvratt, M.J. and A.A. Patchett, 1985. Recent developments in the design of angiotensin-converting enzyme inhibitors. Med. Res. Rev., 5: 483-531.
PubMedDirect Link - Khodadadi, M., M. Nateghpour, E. Souri, L. Farivar, A.M. Haghi, A. Rahimi-Froushani and Z. Karbalaei, 2013. Evaluation of effectiveness of ethanolic extract of artemisia aucheri, individually and in combination with chloroquine, on chloroquine - sensitive strain of Plasmodium berghei in Sourian Mice. Iran. J. Publ. Health, 42: 883-888.
Direct Link - Mishra, N., P. Arora, B. Kumar, L.C. Mishra, A. Bhattacharya, S.K. Awasthi and V.K. Bhasin, 2008. Synthesis of novel substituted 1,3-diaryl propenone derivatives and their antimalarial activity In vitro. Eur. J. Med. Chem., 43: 1530-1535.
CrossRefPubMedDirect Link - Buckner, F.S., N.C. Waters and V.M. Avery, 2012. Recent highlights in anti-protozoan drug development and resistance research. Int. J. Parasitol. Drugs Drug Resist., 2: 230-235.
CrossRefDirect Link - Woodrow, C.J. and S. Krishna, 2006. Antimalarial drugs: recent advances in molecular determinants of resistance and their clinical significance. Cell. Mol. Life Sci., 63: 1586-1596.
CrossRefPubMedDirect Link - Yakasai, A.M., M. Hamza, M.M. Dalhat, M. Bello and M.A. Gadanya et al., 2015. Adherence to artemisinin-based combination therapy for the treatment of uncomplicated malaria: A systematic review and meta-analysis. J. Trop. Med.
CrossRefDirect Link - Nzila, A., M. Rottmann, P. Chitnumsub, S.M. Kiara and S. Kamchonwongpaisan et al., 2010. Preclinical Evaluation of the Antifolate QN254, 5-Chloro- N′6′-(2,5-Dimethoxy-Benzyl)-Quinazoline-2,4,6-Triamine, as an Antimalarial Drug Candidate. Antimicrob. Agents Chemother, 54: 2603-2610.
CrossRefDirect Link - Schunk, M., W.P. Kumma, I.B. Miranda, M.E. Osman and S. Roewer et al., 2006. High prevalence of drug-resistance mutations in Plasmodium falciparum and Plasmodium vivax in southern Ethiopia. Malar. J., Vol. 5.
CrossRefDirect Link - Hovlid, M.L. and E.A. Winzeler, 2016. Phenotypic screens in antimalarial drug discovery. Trends Parasitol., 32: 697-707.
CrossRefDirect Link - Rojas-Aguirre, Y., F. Hernandez-Luis, C. Mendoza-Martinez, C.P. Sotomayor and L.F. Aguilar et al., 2012. Effects of an antimalarial quinazoline derivative on human erythrocytes and on cell membrane molecular models. Biochimica Biophysica Acta (BBA)-Biomembr., 1818: 738-746.
CrossRefDirect Link - Loiseau, P.M. and D.X. Nguyen, 1996. Plasmodium berghei mouse model: antimalarial activity of new alkaloid salts and of thiosemicarbazone and acridine derivatives. Trop. Med. Int. Health, 1: 379-384.
CrossRefPubMedDirect Link - Schluter, K., R.D. Walter, B. Bergmann and T. Kurz, 2006. Arylmethyl substituted derivatives of Fosmidomycin: synthesis and antimalarial activity. Eur. J. Med. Chem., 41: 1385-1397.
CrossRefPubMedDirect Link - Xue, C.X., S.Y. Cui, M.C. Liu, Z.D. Hu and B.T. Fan, 2004. 3D QSAR studies on antimalarial alkoxylated and hydroxylated chalcones by CoMFA and CoMSIA. Eur. J. Med. Chem., 39: 745-753.
CrossRefDirect Link - Valla, A., B. Valla, D. Cartier, R. Le Guillou and R. Labia et al., 2006. New syntheses and potential antimalarial activities of new ‘retinoid-like chalcones’. Eur. J. Med. Chem., 41: 142-146.
CrossRefDirect Link - El Hage, S., M. Ane, J.L. Stigliani, M. Marjorie, H. Vial, G. Baziard-Mouysset and M. Payard, 2009. Synthesis and antimalarial activity of new atovaquone derivatives. Eur. J. Med. Chem., 44: 4778-4782.
CrossRefDirect Link - Nigussie, D., T. Beyene, N.A. Shah and S. Belew, 2015. New targets in malaria parasite chemotherapy: A review. Malar. Control Elimination, Vol. S1.
CrossRefDirect Link - Katsuno, K., J.N. Burrows, K. Duncan, R.H. van Huijsduijnen and T. Kaneko et al., 2015. Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat. Rev. Drug Discov., 14: 751-758.
CrossRefDirect Link - Chen, Y. and R. Xu, 2015. Network-based gene prediction for Plasmodium falciparum malaria towards genetics-based drug discovery. BMC Genomics, Vol. 16.
CrossRefDirect Link - Meyers, M.J. and E.D. Goldberg, 2012. Recent advances in plasmepsin medicinal chemistry and implications for future antimalarial drug discovery efforts. Curr. Top. Med. Chem., 12: 445-455.
PubMedDirect Link - Delves, M., D. Plouffe, C. Scheurer, S. Meister and S. Wittlin et al., 2012. The activities of current antimalarial drugs on the life cycle stages of Plasmodium: A comparative study with human and rodent parasites. PLOS Med., Vol. 9.
CrossRefDirect Link - Li, F., V. Bounkeua, K. Pettersen and J.M. Vinetz, 2016. Plasmodium falciparum ookinete expression of plasmepsin VII and plasmepsin X. Malar. J., Vol. 15.
CrossRefDirect Link - Huizing, A.P., M. Mondal and A.K.H. Hirsch, 2015. Fighting malaria: Structure-guided discovery of nonpeptidomimetic plasmepsin inhibitors. J. Med. Chem., 58: 5151-5163.
CrossRefPubMedDirect Link - Gambini, L., L. Rizzi, A. Pedretti, O. Taglialatela-Scafati and M. Carucci et al., 2015. Picomolar inhibition of plasmepsin v, an essential malaria protease, achieved exploiting the prime region. PLOS ONE, Vol. 11.
Direct Link - Ersmark, K., B. Samuelsson and A. Hallberg, 2006. Plasmepsins as potential targets for new antimalarial therapy. Med. Res. Rev., 26: 626-666.
CrossRefPubMedDirect Link - Berry, C., 1997. New targets for antimalarial therapy: The plasmepsins, malaria parasite aspartic proteinases. Biochem. Educ., 25: 191-194.
CrossRefDirect Link - Hodder, A.N., B.E. Sleebs, P.E. Czabotar, M. Gazdik and Y. Xu, et al., 2015. Structural basis for plasmepsin V inhibition that blocks export of malaria proteins to human erythrocytes. Nat. Struct. Mol. Biol., 22: 590-596.
CrossRefPubMedDirect Link - Sun, Y., J. Wu, J. Wan and M.W. Ding, 2009. Efficient synthesis and fungicidal activities of 2-alkylamino-6-(1H-1,2,4-triazol-1-yl)-thieno[2,3-d]pyrimidin-4(3H)-ones. Arch. Org. Chem., 2009: 111-118.
CrossRefDirect Link - Edafiogho, I.O., M.G. Qaddoumi, K.V.V. Ananthalakshmi, O.A. Phillips and S.B. Kombian, 2014. Synthesis, neuronal activity and mechanisms of action of halogenated enaminones. Eur. J. Med. Chem., 76: 20-30.
CrossRefDirect Link - Kabri, Y., N. Azas, A. Dumetre, S. Hutter and M. Laget et al., 2010. Original quinazoline derivatives displaying antiplasmodial properties. Eur. J. Med. Chem., 45: 612-622.
CrossRefPubMedDirect Link - Jafari, E., M.R. Khajouei, F. Hassanzadeh, G.H. Hakimelahi and G.A. Khodarahmi, 2016. Quinazolinone and quinazoline derivatives: Recent structures with potent antimicrobial and cytotoxic activities. Res. Pharm. Sci., 11: 1-14.
Direct Link - Sutherland, J.B., T.M. Heinze, L.K. Schnackenberg, J.P. Freeman and A.J. Williams, 2011. Biotransformation of quinazoline and phthalazine by Aspergillus niger. J. Biosci. Bioeng., 111: 333-335.
CrossRefDirect Link - Sharma, P.C., G. Kaur, R. Pahwa, A. Sharma and H. Rajak, 2011. Quinazolinone analogs as potential therapeutic agents. Curr. Med. Chem., 18: 4786-4812.
PubMedDirect Link - Asif, M., 2014. Chemical characteristics, synthetic methods, and biological potential of quinazoline and quinazolinone derivatives. Int. J. Med. Chem.
CrossRefDirect Link - Maarouf, A.R., E.R. El-Bendary and F.E. Goda, 2004. Synthesis and evaluation of some novel quinazolinone derivatives as diuretic agents. Arch. Pharm. (Weinheim), 337: 527-532.
CrossRefPubMedDirect Link - Sharma, M., S. Pandey, K. Chauhan, D. Sharma, B. Kumar and P.M.S. Chauhan, 2012. Cyanuric chloride catalyzed mild protocol for synthesis of biologically active dihydro/spiro quinazolinones and quinazolinone-glycoconjugates. J. Org. Chem., 77: 929-937.
CrossRefDirect Link - Selvam, T.P. and P.V. Kumar, 2011. Quinazoline marketed drugs-a review. Res. Pharm., 1: 1-21.
Direct Link - Santelli-Rouvier, C., B. Pradines, M. Berthelot, D. Parzy and J. Barbe, 2004. Arylsulfonyl acridinyl derivatives acting on Plasmodium falciparum. Eur. J. Med. Chem., 39: 735-744.
CrossRefPubMedDirect Link - Alafeefy, A.M., A.A. Kadi, A.S. El-Azab, S.G. Abdel-Hamide and M.H.Y. Daba, 2008. Synthesis, analgesic and anti-inflammatory evaluation of some new 3H-quinazolin-4-one derivatives. Arch. Pharm. (Weinheim), 341: 377-385.
CrossRefDirect Link - Bhattacharjee, A.K., M.G. Hartell, D.A. Nichols, R.P. Hicks, B. Stanton, J.E. van Hamont and W.K. Milhous, 2004. Structure-activity relationship study of antimalarial indolo [2,1-b]quinazoline-6,12-diones (tryptanthrins). Three dimensional pharmacophore modeling and identification of new antimalarial candidates. Eur. J. Med. Chem., 39: 59-67.
CrossRefPubMedDirect Link - Galli, U., L. Lazzarato, M. Bertinaria, G. Sorba, A. Gasco, S. Parapini and D. Taramelli, 2005. Synthesis and antimalarial activities of some furoxan sulfones and related furazans. Eur. J. Med. Chem., 40: 1335-1340.
CrossRefPubMedDirect Link - Pandey, S.K., A. Singh, A. Singh and Nizamuddin, 2009. Antimicrobial studies of some novel quinazolinones fused with [1,2,4]-triazole, [1,2,4]-triazine and [1,2,4,5]-tetrazine rings. Eur. J. Med. Chem., 44: 1188-1197.
CrossRefPubMedDirect Link - Grover, G. and S.G. Kini, 2006. Synthesis and evaluation of new quinazolone derivatives of nalidixic acid as potential antibacterial and antifungal agents. Eur. J. Med. Chem., 41: 256-262.
CrossRefPubMedDirect Link - Jatav, V., P. Mishra, S. Kashaw and J.P. Stables, 2008. CNS depressant and anticonvulsant activities of some novel 3-[5-substituted 1,3,4-thiadiazole-2-yl]-2-styryl quinazoline-4(3H)-ones. Eur. J. Med. Chem., 43: 1945-1954.
CrossRefDirect Link - Al-Obaid, A.M., S.G. Abdel-Hamide, H.A. El-Kashef, A.A.M. Abdel-Aziz, A.S. El-Azab, H.A. Al-Khamees and H.I. El-Subbagh, 2009. Substituted quinazolines, part 3. Synthesis, In vitro antitumor activity and molecular modeling study of certain 2-thieno-4(3H)-quinazolinone analogs. Eur. J. Med. Chem., 44: 2379-2391.
CrossRefPubMedDirect Link - Giri, R.S., H.M. Thaker, T. Giordano, J. Williams, D. Rogers, V. Sudersanam and K.K. Vasu, 2009. Design, synthesis and characterization of novel 2-(2,4-disubstituted-thiazole-5-yl)-3-aryl-3H-quinazoline-4-one derivatives as inhibitors of NF-κB and AP-1 mediated transcription activation and as potential anti-inflammatory agents. Eur. J. Med. Chem., 44: 2184-2189.
CrossRefPubMedDirect Link - Kumar, V., A. Mahajan and K. Chibale, 2009. Synthetic medicinal chemistry of selected antimalarial natural products. Bioorg. Med. Chem., 17: 2236-2275.
CrossRefPubMedDirect Link - Kaur, K., M. Jain, T. Kaur and R. Jain, 2009. Antimalarials from nature. Bioorg. Med. Chem., 17: 3229-3256.
CrossRefDirect Link - Jain, V., M. Yogavel, Y. Oshima, H. Kikuchi, B. Touquet, M.A. Hakimi and A. Sharma, 2015. Structure of Prolyl-tRNA synthetase-halofuginone complex provides basis for development of drugs against malaria and toxoplasmosis. Structure, 23: 819-829.
CrossRefPubMedDirect Link - Kikuchi, H., S. Horoiwa, R. Kasahara, N. Hariguchi, M. Matsumoto and Y. Oshima, 2014. Synthesis of febrifugine derivatives and development of an effective and safe tetrahydroquinazoline-type antimalarial. Eur. J. Med. Chem., 76: 10-19.
CrossRefPubMedDirect Link - McLaughlin, N.P., P. Evans and M. Pines, 2014. The chemistry and biology of febrifugine and halofuginone. Bioorg. Med. Chem., 22: 1993-2004.
CrossRefPubMedDirect Link - Zhu, S., G. Chandrashekar, L. Meng, K. Robinson and D. Chatterji, 2012. Febrifugine analogue compounds: synthesis and antimalarial evaluation. Bioorg. Med. Chem., 20: 927-932.
CrossRefPubMedDirect Link