
Mohammad Hadi Goharbari
Department of Toxicology and Pharmacology, Faculty of Pharmacy and Pharmaceutical Sciences Research Center,
Tehran University of Medical Sciences, Tehran, 1417614411, Iran
Amir Shadboorestan
Department of Toxicology and Pharmacology, Faculty of Pharmacy and Pharmaceutical Sciences Research Center,
Tehran University of Medical Sciences, Tehran, 1417614411, Iran
Mohammad Abdollahi
Department of Toxicology and Pharmacology, Faculty of Pharmacy and Pharmaceutical Sciences Research Center,
Tehran University of Medical Sciences, Tehran, 1417614411, Iran
LiveDNA: 98.1833
International Journal of Pharmacology
Year: 2016 | Volume: 12 | Issue: 3 | Page No.: 249-261







ABSTRACT
Oxidative stress condition which is due to increased cellular free radicals has a main role in cell injury. Although free radicals are generated in normal metabolism, they may also be produced in excessive amounts during pathological conditions such as oxygen toxicity, drug overdose, chemical toxicity, ischemic-hypoxic injury and so on. The main parts of free radicals are Reactive Oxygen Species (ROS) which are mainly generated in cellular respiration. Increase in amount of ROS in pathological conditions can disturb mitochondrial function and make cellular damage. Thyroid hormones can act as a cytoprotective and antioxidant by inducing and activating some defense mechanisms against increased free radicals and mitochondrial oxidative stress. To review and categorize underlying mechanisms for the inhibitory effects of thyroid hormones on mitochondrial oxidative stress, Medline, Scopus and Web of science were searched for in vitro , in vivo, animal and human studies reporting antioxidant and cytoprotective effect of thyroid hormones in oxidative stress. After excluding duplicate, irrelevant and old articles the studies which had eligible criteria from 1980-2015 were included. Fifty one studies were included and evaluated in our study. It was found that thyroid hormones can induce cytoprotective and mitochondrial antioxidant effects through three mechanisms, consisting of increased activity and expression of uncoupling proteins, increased activity of mitoKATP channels, and increased activity and expression of antioxidant enzymes. Thyroid hormones have antioxidant effects through different mechanisms. More studies are needed to determine probable further mechanisms for thyroid hormones antioxidant effects and to confirm tissue hypothyroidism in oxidative stress.
PDF Abstract XML References Citation
How to cite this article
Mohammad Hadi Goharbari, Amir Shadboorestan and Mohammad Abdollahi, 2016. Inhibitory Effects of Thyroid Hormones on Mitochondrial
Oxidative Stress: A Systematic Review. International Journal of Pharmacology, 12: 249-261.
DOI: 10.3923/ijp.2016.249.261
URL: https://scialert.net/abstract/?doi=ijp.2016.249.261
DOI: 10.3923/ijp.2016.249.261
URL: https://scialert.net/abstract/?doi=ijp.2016.249.261
INTRODUCTION
Cell injury by free radicals, particularly Reactive Oxygen Species (ROS) is one of the main mechanisms for cell damages. Free radicals are atoms or molecules having single unpaired electron in the outer orbital. Energy created by this unstable configuration may lead to chemical reaction with cellular key molecules (carbohydrate, lipids, proteins and nucleic acids which are the main components of cells) initiating autocatalytic reactions. Free radicals are generated during normal metabolism and in most pathological conditions such as oxygen toxicity, ionization radiation, ultraviolet light, drugs, chemicals, toxins, reperfusion after ischemic injury (Droge, 2002; Hensely et al., 2000; Li and Jackson, 2002; Salvemini and Cuzzocrea, 2002). The ROS are molecules derived from oxygen and produced in some normal cellular activities such as cellular respiration, however, they may be generated in excessive amounts in pathological conditions and make harmful effects on the cells. Superoxide anion (O2–), hydrogen peroxide (H2O2), hydroxyl radical (•HO) are the principal ROS involved in the cell injury (Droge, 2002). In a review study, Saeidnia and Abdollahi (2013) thoroughly demonstrated the role of ROS in cellular damage and Oxidative Stress Related Diseases (OSRDs). The cells can control the ROS concentration by some defense mechanisms to prevent damage. When there is ROS overproduction or ineffective cellular defense, free radicals accumulate and their concentration increases. The result is an excess of free radicals, leading to a pathologic condition called oxidative stress. In other words, oxidative stress is an accumulation of oxygen derived free radicals, which have an important role in a wide variety of pathologic processes and diseases, including cancer, Alzheimer’s disease, aging and so on (Abdollahi et al., 2014). Free radicals are degraded by: (1) Intracellular enzymes such as superoxide dismutase (SOD), catalase, glutathione peroxidase, (2) Endogenous or exogenous antioxidants such as vitamin A, vitamin C, vitamin E, cysteine, glutathione, selenium, ceruloplasmin and transferrin and (3) Spontaneous decay. Superoxide anion (O2–) is a precursor for most ROS. In dismutation reaction superoxide anion is converted to hydrogen peroxide (H2O2) by SOD. After that, the majority of hydrogen peroxide is reduced to water and a small amount converted to hydroxyl radical (•HO). Electron transportation in cellular respiration is the main process to generate ROS in the most tissues. In this process, some electrons may leak and reduce oxygen molecules which result in producing ROS (Turrens, 2003). It is very important to know, superoxide formation is related to two factors: (1) Concentration of electron donors and (2) Concentration of oxygen (Szewczyk et al., 2009).
It is clear that any factor which can inhibit or control oxidative stress may have antioxidant and cytoprotective effects. With due attention to the important role of thyroid hormones in the cell biology, the protective effects of these hormones on the oxidative stress condition have been studied in the recent years. The role of thyroid hormones as an oxidant or antioxidant is controversial. Some studies indicated thyroid hormone-induced oxidative stress (Venditti and di Meo, 2006), whereas in a recent animal research by Abdolghaffari et al. (2015), the cardioprotective effects of triiodothyronine against phosphine-induced cardiac and mitochondrial toxicity have been confirmed. They administrated T3 at 3 doses (1, 2 and 3 μg kg–1) in a rat model of aluminum phosphate induced cardiotoxicity and found that T3 at the dose of 3 μg kg–1 significantly improved ECG and oxidative stress parameters (Abdolghaffari et al., 2015). There are some other studies showing the antioxidant and cytoprotective effects of thyroid hormones in oxidative stress condition (De Castro et al., 2014; Forini et al., 2011, 2015; Mourouzis et al., 2013; Pantos and Cokkinos, 2010; Pantos et al., 2010a, b, 2011a, b, 2007, 2009). In contrary conditions, these questions arise: What is the role of thyroid hormones in oxidative stress condition? Do thyroid hormones have antioxidant and cytoprotective effects? What are the mechanisms for these effects?
In trying to make a clearance on such controversies and the answer to the above questions, in this review article, the antioxidant effects of thyroid hormones and mechanisms related to these effects have been criticized.
METHODS
This study was performed according to the recommendations of the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement (Moher et al., 2009).
Eligibility criteria, information sources and search protocol:
In this study, electronic search was performed on Medline, Scopus and Web of science. All in vitro, in vivo, animal and human studies reporting antioxidant and cytoprotective effects of thyroid hormones in oxidative stress were included. Additional studies were identified through other sources and the records screened with titles and abstracts. After excluding duplicate, irrelevant and old articles, the studies which showed the antioxidant effects of T3 and described the mechanisms of these effects from 1980-2015 were evaluated.
Study selection and quality assessment: Assessment of the studies was based on their title or abstract and those studies which had the eligibility criteria were selected for full text review.
Data collection, data items and synthesis of results: Data were collected and classified according to the thyroid hormones inhibitory effects on mitochondrial oxidative stress. Free radicals, oxidative stress, antioxidant enzymes and the mechanisms of the thyroid hormones on oxidative stress were discussed. The mechanisms of the thyroid hormones inhibitory effect on mitochondrial oxidative stress were categorized and discussed in three sections based on the data and the results of the selected articles.
RESULTS
Study selection and characteristics: Our initial search through database identified 138 published articles; on the other side, we had 19 additional records through other sources. After removing duplicate articles, 152 articles remained. Titles and abstracts were reviewed and 87 papers were excluded. The full texts of the remaining 65 articles were assessed for eligibility. Nine papers had old data, two papers had duplicated data and three papers were irrelevant that all of them were excluded. Finally 51 articles were included. Figure 1 shows the flow chart of study selection.
Results of studies: In our full data extraction, the main mechanisms for the inhibitory effects of thyroid hormones on mitochondrial oxidative stress can be categorized and summarized as follows: (1) Increased activity and expression of uncoupling proteins, (2) Increased activity of mitoKATP channels (ATP-sensitive potassium channels) and (3) Increased activity and expression of antioxidant enzymes.
Increased activity and expression of uncoupling proteins: Uncoupling proteins are located in the mitochondrial inner membranes and have a major duty in cellular physiology. These proteins are effective in maintaining the proton (H+) gradient on both sides of the inner membrane and transport proton (H+) from inter-membrane space to the mitochondrial matrix. In other words uncoupling proteins (UCPs) are the number of mitochondrial anion carrier proteins which transport anion from the mitochondrial matrix to inter-membrane space and proton from inter-membrane space to the mitochondrial matrix (Nedergaard et al., 2005). This class of mitochondrial proteins has five known types including UCP1, UCP2, UCP3, UCP4 and UCP5. The amounts and expression of uncoupling proteins are regulated by numerous factors such as thyroid hormones, norepinephrine, epinephrine and leptin (Gong et al., 1997).
It looks each UCP has a particular function and the expression of UCPs varies in different tissues according to necessity. For example, UCP1 called thermogenin is responsible for non–shivering thermogenesis and it is more expressed in Brown Adipose Tissue (BAT), whereas, UCP2 are further expressed in muscle cells and they are responsible for the elimination of ROS in oxidative stress condition (Arsenijevic et al., 2000).
It should be noted that cellular respiration is always associated with the production of small amounts of ROS (Raha and Robinson, 2000). Increased ROS amounts are very harmful to cells. There are some mechanisms for eliminating and controlling ROS amounts to prevent cells from ROS adverse effects.
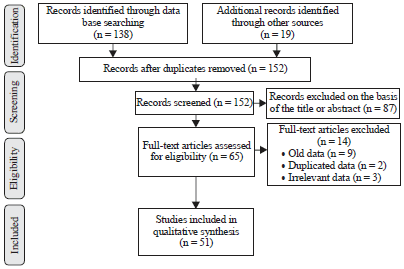 | |
| Fig. 1: | Flow chart of study selection |
Mild uncoupling is one of these mechanisms. Mild uncoupling is done by UCPs that transport anions and cations and is mediated by thyroid hormones in mitochondria (Skulachev, 1996, 1998; Starkov, 1997).
As mentioned, UCPs transport proton from mitochondrial inter-membrane space to mitochondrial matrix and transport anions vice versa. By this mechanism, UCPs reduce concentration of ROS and protect cells from lipid peroxidation (Nedergaard et al., 2005; Ricquier and Bouillaud, 2000).
The UCPs have a main role in the proton-leak process in mitochondria. There are 2 kinds of proton-leak in mitochondria: (1) Basal, it occurs in all tissues and there is no known regulatory mechanism for it and (2) Inducible, this kind of proton-leak process is done by UCPs and carefully regulated by known regulatory mechanism of Brand et al. (1999). Negre-Salvayre et al. (1997) showed the role of UCPs particularly UCP2 in reduction of ROS and prevention of oxidative stress damages. One year later, Skulachev (1998) showed that mild uncoupling is a defense mechanism in cellular respiration and UCPs act as an antioxidant. Vidal-Puig et al. (2000) represented that proton-leak was reduced and ROS production was increased in UCP3 knockout mice. Some studies demonstrated UCP2 and UCP3 are more effective as an antioxidant in ROS controlling than the other UCPs (Arsenijevic et al., 2000; Negre-Salvayre et al., 1997; Vidal-Puig et al., 2000). Dulloo and Samec (2001) showed UCP1 has a damper effect on ROS productions. Ramsden et al., 2012 found UCP4 and UCP5 reduced superoxide anion formation in neurons of Parkinson’s patients. In this way they showed neuroprotective effects of UCPs against neurotoxins and ROS. The UCPs can restrict harmful effects of ROS on macrophage and prevent cellular degeneration (Rousset et al., 2004). The specificities of the known five types of UCPs are shown in Table 1.
Regulation of mitochondrial DNA transcription is one of the important effects of thyroid hormones. Considering this effect, thyroid hormone can influence mitochondrial RNAs and protein concentration (Cioffi et al., 2013; Wrutniak-Cabello et al., 2001). In a study, in hypothyroid rats, mtRNA concentration raised after T3 administration. Hypothyroidism was induced in rats by tapazol, for 4-5 weeks and after that T3 was administrated at a dose 20 μg/100 g/day. Steady concentration of mtRNA in hepatocytes was 2-8 times higher after administration of T3 in comparison with when T3 was not administrated, whereas there was no significant change in mitochondrial rRNA concentration (Mutvei et al., 1989).
Tri-idothyronine can induce increasing of uncoupling proteins. In some animal studies, UCP2 and UCP3 expression was increased by T3 administration in cardiomyocytes and skeletal muscle cells (Gong et al., 1997; Lanni et al., 1999, 1997; Silvestri et al., 2005).
The UCPs (particularly UCP2 and UCP3) act as an antioxidant in oxidative stress condition and are effective in reduction of free radicals. In addition, expression and function of UCPs are increased by thyroid hormones. So thyroid hormones are critical in regulating and controlling free radicals in oxidative stress condition (Lanni et al., 2003). It is very important to know, in spite of increased expression and function of UCPs of thyroid hormones, ATP production increases. In an animal study on rat, after T3 administration, despite high UCPs expression ATP generation was increased during oxidative stress (Short et al., 2001). Summary of studies related to the antioxidant effects of UCPs and thyroid hormones is shown in Table 2.
Increased activity of mitoKATP channels: Potassium channels are a class of ion channels which exist in almost all human cells. They are classified according to their functions and site of action. There are 4 potassium channel classes: (1) Calcium-activated potassium channel, (2) Inwardly rectifying potassium channel, (3) Tandem pore domain potassium channel and (4) Voltage-gated potassium channel (Jessell, 2000; Littleton and Ganetzky, 2000).
| Table 1: | Specificities of UCPs |
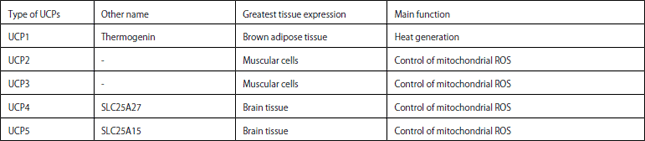 | |
| UCP1: Uncoupling protein 1, UCP2: Uncoupling protein 2, UCP3: Uncoupling protein 3, UCP4: Uncoupling protein 4, UCP5: Uncoupling protein 5, ROS: Reactive oxygen species | |
| Table 2: | Effects of uncoupling proteins and thyroid hormones on ROS and oxidative stress |
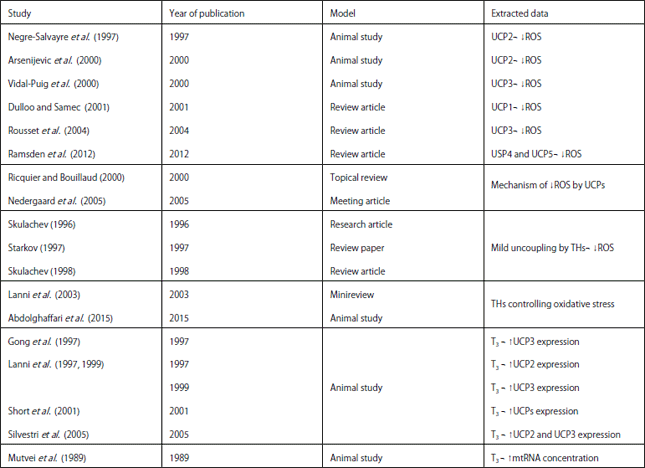 | |
| UCP: Uncoupling protein, UCP1: Uncoupling protein 1, UCP2: Uncoupling protein 2, UCP3: Uncoupling protein 3, UCP4: Uncoupling protein 4, UCP5: Uncoupling protein 5, ROS: Reactive oxygen species, TH: Thyroid hormone, T3: Triiodothyronine, mtRNA: Mitochondrial RNA | |
Inwardly rectifying potassium channels have 7 subfamilies (Kir-1-Kir-7) and mitochondrial ATP-sensitive potassium channel is one of them (Kubo et al., 2005; Suzuki et al., 1997; Zhou et al., 1999). The ATP-sensitive potassium channels were first identified in myocardial cells by a Japanese research group (Noma, 1983). This class of potassium channels regulates cellular and intracellular organelles membrane potassium concentration and has an important function in cellular biologic activities consisting of cell metabolism, apoptosis and gene expression. According to function and site of action, this class is categorized into three subclasses: (1) Sarcolemmal (sarcKATP), (2) Mitochondrial (mitoKATP) and (3) Nuclear (nuclKATP) (Zhuo et al., 2005). The MitoKATP channels were first identified in liver cell mitochondria by Inoue et al. (1991). After that, these subclasses of potassium channels were identified in mitochondria of heart, brain, kidney, musculoskeletal and other human cells, respectively (Bajgar et al., 2001; Cancherini et al., 2003; Debska et al., 2002, 2001).
The cytoprotective effect of mitoKATP channels in neural and cardiovascular damages has been shown in several studies (Busija et al., 2008; Szewczyk et al., 2009). The cardioprotective effect of mitoKATP channels in ischemic-hypoxic conditions play an important role against oxidative stress. Activation of these channels in ischemic damages can protect cardiomyocytes against further injuries. In this process, sarcKATP and mitoKATP have a dominant role (Liu et al., 1998; Zhuo et al., 2005).
Ordinarily, mitochondrial function decreases at the beginning of the cellular energy crisis. This is due to mitochondrial internal membrane potential disturbances and trans-membrane ion transport imbalance which may lead to free radicals overproduction (Zhuo et al., 2005). In this condition, mitoKATP channels are activated and help the cell to regulate membrane potential, intracellular Ca+2 concentration and ATP production, as well as reduction of free radical concentration (Xu et al., 2001).
Akao et al. (2001) showed the role of mitoKATP channels against cell death in oxidative stress. They emphasized mitoKATP channels have an important function in preserving mitochondrial integrity. Ardehali (2004) showed that mitochondrial ATP-binding cassette protein 1 (mABC1) has a beneficial effect on cell protection against oxidative stress by activation of mitoKATP.
It seems, KATP channels have an important activity in ischemic preconditioning (IPC). With knockout of sarcKATP genes, myocardial damage was increased in ischemic condition (Suzuki et al., 2002). Preconditioning phenomenon was first described by Murry et al. (1986). In this phenomenon, after short term ischemia-hypoxia episodes, myocardial cells prepare to deal with prolonged ischemia attacks. The IPC leads to a decrease in the size of infarction and reduces the risk of arrhythmias (Cohen et al., 2000; Kloner, 1998; Murry et al., 1986). Increased ROS concentration is a trigger to open the KATP channels in IPC. In other words, when the ROS concentration is rising, KATP channels will be opened to reduce the ROS amounts and their harmful effects (Zhang et al., 2001).
There are three hypotheses regarding cardioprotective effects of the KATP channels: (1) Decrease in the mitochondrial Ca+2uptake, (2) Swelling of the mitochondrial matrix and increase in ATP synthesis and (3) Decrease in ROS levels (Ardehali et al., 2005). These three hypotheses are shown in Fig. 2. Diazoxide and pinacidil effects on decreasing mitochondrial Ca2+ uptake is due to activation of mitoKATP channels. These effects are reversible by a channel blocker such as 5-HD (Holmuhamedov et al., 1999). Murata et al. (2001) showed reduction of mitochondrial calcium overload is a consequence of partial mitochondrial membrane depolarization by mitoKATP channels. They presented this process is a significant mechanism of cardioprotective effect of mitoKATP channels. Opening of mitoKATP channels leads to mitochondrial matrix swelling and this process results in electron transport chain activation and generating more ATP, which helps myocardial recovery (Grover and Garlid, 2000; Halestrap, 1989; O'Rourke, 2000). In other words, mitochondrial matrix expansion results in improving fatty acid oxidation, cellular respiration and ATP production. Increased ATP production following mitoKATP channel activation was shown in the other studies (Kowaltowski et al., 2001).
Reactive oxygen species production has increased in the early IPC period and leads to mitoKATP channel activation. Opening of mitoKATP channels has an important role in the reduction of ROS concentration in both early and delayed phases of IPC and it results in preventing further myocardial damages (Forbes et al., 2001; O'Rourke, 2004; Ozcan et al., 2002; Hoek et al., 1998). Likewise, it can be noted that opening of mitoKATP channels has an important task in decreasing oxidative stress damages. This process is complementary for mild uncoupling to reduce mitochondrial ROS concentration (Skulachev, 1996). Heinen et al. (2007) and Kulawiak et al. (2008) confirmed that opening of mitoKATP channels in oxidative stress leads to reduction of ROS concentration.
The effect of thyroid hormones on potassium channels has been studied previously. These channels can be more activated in the presence of thyroid hormones. Sakaguchi et al. (1996) considered the effects of T3 on cardiac electrophysiology in guinea pig. They found that inward rectifier potassium channels become more active with T3 administration. In addition, thyroid hormones have a regulatory effect on mRNA expression of potassium channels. Abe et al. (1998) indicated this effect of thyroid hormones on cardiomyocytes of rat in 1998 and Nishiyam et al. (1998) confirmed the regulatory effects of thyroid hormones on potassium channels isolated cardiomyocytes from euthyroid rat and exposed these cells to H2O2 and found that with T3 administration mitoKATP channels were becoming more active and the effect of H2O2 as a free radical was decreased on cardiomyocyte (Forini et al., 2011).
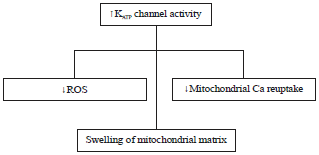 | |
| Fig. 2: | Three hypotheses for cytoprotective efffects of KATP channels |
| Table 3: | Cytoprotective effects of thyroid hormones via increased activity of mitoKATP channels |
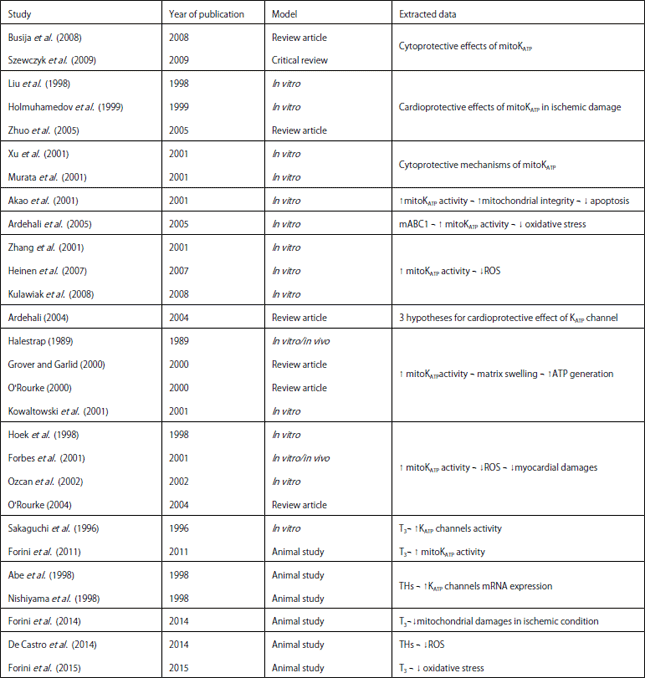 | |
| MitoKATP: Mitochondrial ATP-sensitive potassium channel, mABC1: Mitochondrial ATP-binding cassette protein 1, ROS: Reactive oxygen species, K ATP channel: ATP-sensitive potassium channel, ATP: Adenosine triphosphate, T3: Triiodothyronine, TH: Thyroid hormone | |
In the other study, Forini et al. (2014) considered the cardioprotective effects of T3 in ischemic condition. They showed T3 treatment can prevent further mitochondrial disturbance and damage in ischemic cases. Forini et al. (2015) thoroughly described T3 cardioprotective effects, particularly on the oxidative stress condition in a review article. De Castro et al., (2014) showed the effects of thyroid hormones on reduction of ROS concentration and also control of oxidative stress in infected rat myocardium. Summary of studies related to the antioxidant effects of mitoKATP channels and thyroid hormones is shown in Table 3.
Increased activity and expression of antioxidant enzymes: One of the important mechanisms for reducing the amounts of free radicals is increased activity and expression of the enzymes catalyzing free radicals to low risk or harmless products. Superoxide dismutase (SOD), catalase and glutathione peroxidase are the most important antioxidant enzymes which catalyze free radicals.
Superoxide dismutase (SOD): Superoxide dismutase enzymes are a group of enzymes catalyzing the dismutation reaction of superoxide (O2–) to oxygen or hydrogen peroxide (H2O2). Superoxide anion (O2–) is a byproduct of oxygen metabolism. If the effects of superoxide are not controlled, it can be harmful to cells. For this reason, superoxide dismutase enzymes have an important cytoprotective activity against harmful effects of superoxide. Large amounts of hydrogen peroxide are harmful to cells, but these harmful effects are much less than superoxide effects and hydrogen peroxide is immediately degraded by some enzymes such as catalase.
The SOD has a critical activity in living cells exposed to oxygen. Dismutation reactions by SOD are described below:
Enzymatic activity of SOD was first identified by McCord and Fridovich (1968). Before that time, SODs had known as metalloproteins with unknown function. SOD enzymes have a polypeptide structure that in their active sites has metals such as copper, zinc, manganese, iron or nickel as a co-factor. They are classified according to their metal and protein fold: (1) Copper and zinc type (Cu/Zn type), which binds both copper and zinc, (2) Iron or manganese type (Fe or Mn type), which binds either iron or manganese and (3) Nickel type (Ni type), which binds nickel. All of them are exist in the human cells. In oxidative stress condition, the activity and concentration of SODs increases and they play their antioxidant activity by reducing superoxide concentration.
Catalase: Catalase is one of the important enzymes in all living cells exposed to oxygen. Catalase acts as a cytoprotective agent against ROS in oxidative stress condition. This enzyme catalyzes oxygen peroxide to oxygen and water:
Catalase is a tetramer with 4 polypeptide chains that each one has 500 aminoacids. The H2O2 is a byproduct of oxygen metabolism, which can be harmful to cells in oxidative stress condition. For this reason, it should be rapidly converted to harmless products.
Glutathione peroxidase (GPx): Glutathione peroxidase is a general name for an enzyme group having peroxidase activity. They protect cells from oxidative damages. There are 8 known isoenzymes for GPx named GPx-1-GPx-8. GPx catalyzes the hydrogen peroxide to harmless products in this reaction:
If GPx activity is decreased, hydrogen peroxide concentration will be raised and results in lipid and hydrogen peroxidation (Arthur, 2000; Meyer et al., 1994; Yu and Chung, 2006).
It appears that thyroid hormones can increase expression and activity of antioxidant enzymes. Das and Chainy (2001) studied T3 effects on oxidative stress parameters in mitochondria of rat liver cells and found H2O2 concentration which had risen in hypothyroid state, returned to normal value after T3 administration. They also showed catalase activity was reduced in hypothyroidism, but it was increased and returned to normal value after T3 treatment. In another study on adult rat brain, it was shown antioxidant enzyme activity is influenced by thyroid state. In this way, they indicated antioxidant activities in mitochondria are under the thyroid hormones control and regulation of antioxidant activities and ROS balance are dependent on the normal thyroid function (Das and Chainy, 2004). Choudhury et al. (2003) showed T3 administration to PTU-treated rats (hypothyroid rat) resulted in a rise in the level of catalase activity. In a research study on a normal (euthyroid) fish Anabas testudineus, short term effect of T3 on the enzymes catalyzing ROS was tested. They showed 30 min after T3 injection at a dose of 200 ng in euthyroid fish, lipid peroxidation products such as malondealdehyde were decreased. Triiodothyronine in vitro with concentration of 10-6 M could increase catalase, glutathione peroxidase and glutathione reductase activities as well as glutathione level. They suggest short term administration of T3 has a rapid regulatory effect on the removal of ROS (Sreejith and Oommen, 2006). In a human study done by Naazeri et al. (2014) increased catalase and SOD activities were considered in hyperthyroid state, however, they believe this increased enzyme activity is in response to increased ROS due to hyperthyroid state. Summary of studies related to antioxidant enzymes and thyroid hormones is shown in Table 4.
| Table 4: | Effects of thyroid hormones on antioxidant enzymes |
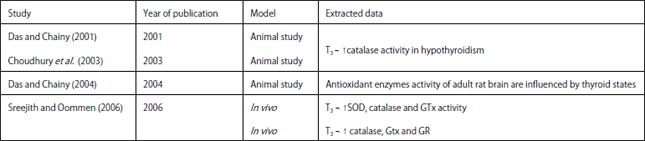 | |
| T3: Triiodothyronine, ROS: Reactive oxygen species, SOD: Superoxide dismutase, GTx: Glutathione peroxidase, GR: Glutathione reductase | |
DISCUSSION
As a result of our study, there are numerous studies addressing antioxidant effects of thyroid hormones and underling mechanisms. Thyroid hormones can have a major role in oxidative stress condition as an antioxidant and cytoprotective agent. It appears cytoprotective effects of thyroid hormones are not only in hypothyroid state, but in euthyroid cases (Sreejith and Oommen, 2006). In euthyroid oxidative stress condition, we may have a tissue hypothyroidism worsening the condition and the positive effects of thyroid hormone administration in this situation are due to improving the tissue hypothyroidism and reduction of ROS concentration. The inhibitory effects of thyroid hormones on mitochondrial oxidative stress are through three mechanisms showed and summarized in Fig. 3.
Uncoupling proteins: One of the significant defense factors of cell to counteract mitochondrial free radicals is uncoupling protein's activity (Arsenijevic et al., 2000; Dulloo and Samec, 2001; Negre-Salvayre et al., 1997; Ramsden et al., 2012; Rousset et al., 2004; Vidal-Puig et al., 2000). As mentioned UCPs transfer protons from mitochondrial intermembrane space to mitochondrial matrix and by this way UCPs can reduce free radicals concentration, when ROS are overloaded during pathological conditions (Nedergaard et al., 2005; Ricquier and Bouillaud, 2000). Thyroid hormones have a critical role in controlling UCPs function and mild uncoupling process (Skulachev, 1996, 1998, Starkov, 1997). In mild uncoupling the activity of UCPs is promoted to decrease ROS concentration, but ATP generation is not disturbed. In other words, the balance between uncoupling process and ATP generation, carried out by thyroid hormones to restore normal cellular function. There are some questions regarding this situation that may guide to further researches: Is there any tissue or cellular hypothyroidism in critical cellular conditions? Do cells need more thyroid hormone to restore their function in critical condition? If there is a cellular hypothyroidism, is it due to decreased thyroid hormones or decreased tissue response to thyroid hormones?
MitoKATP channels: Potassium ATP sensitive channels are a class of ion channels having a critical role in cellular function. Increased mitoKATP channel activity during pathological conditions can lead to decreased mitochondrial calcium overload and ROS concentration. In this way cellular apoptosis and oxidative stress may be decreased. On the other side opening of mitoKATP channels result in swelling of mitochondrial matrix and increases ATP generation (Ardehali, 2004; Kowaltowski et al., 2001; Murata et al., 2001; Xu et al.,2001). So, mitoKATP channel activity has a fundamental role in restoring mitochondrial function during pathological processes (Ardehali, 2004; Szewczyk et al., 2009). Numerous studies showed mitoKATP channel activity is under the control of thyroid hormones (Abe et al., 1998; Forini et al., 2011; Nishiyama et al., 1998; Sakaguchi et al., 1996). Undoubtedly, the function of these channels is disturbed in hypothyroidism, but ambiguity in this regard, is what was discussed in relation to the uncoupling proteins and cellular hypothyroidism. Evaluation of mitoKATP channels functions in different pathologic conditions and the role of thyroid hormones in these conditions are issues requiring further studies.
Antioxidant enzymes: The effect of thyroid hormones on antioxidant enzymes is a controversial topic. The activity of catalase is obviously increased by thyroid hormones in both hypothyroidism and euthyroidism (Choudhury et al., 2003; Das and Chainy, 2001, 2004; Sreejith and Oommen, 2006). The SOD activity was raised after T3 administration in some studies (Sreejith and Oommen, 2006); however, it was decreased in some other studies after T3 administration (Abdolghaffari et al.,2015). In other words, the effect of thyroid hormones on SOD activity in oxidative stress needs to be more studied in further studies. In relation to GTx the issue is similar to SOD.
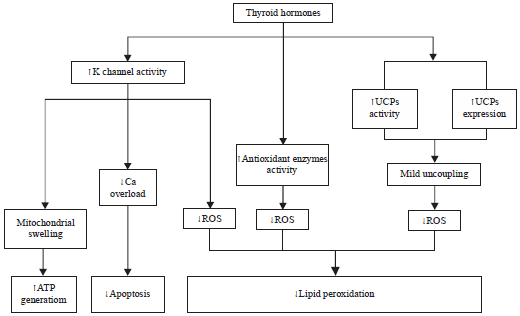 | |
| Fig. 3: | Antioxidant effects of thyroid hormones |
CONCLUSION
All these considerations, let us to place great emphasis on the thyroid hormone effects of oxidative stress, however, there are some questions in this regard which should be answered and need further studies for investigation. For instance, we certainly believe that thyroid hormone by having such effects can be helpful in reducing cardiotoxicity of compounds that act through oxidative stress such as phosphides in the human level. It can also conclude that by use of in-silico phannaco-toxicology system, there will be possible to develop medicines better than thyroid hormones for other oxidative stress diseases. Without a doubt, the role of thyroid hormones in cell biology and body physiology is very important and this role should be further studied during pathological state such as oxidative stress especially in ICU patients.
ACKNOWLEDGMENT
All authors have contributed equally in whole study. Authors wish to thank Iran National Science Foundation and Tehran University of Medical Sciences. All authors have read and approved the final version of the article.
REFERENCES
- Abdolghaffari, A.H., A. Baghaei, R. Solgi, M. Gooshe and M. Baeeri et al., 2015. Molecular and biochemical evidences on the protective effects of triiodothyronine against phosphine-induced cardiac and mitochondrial toxicity. Life Sci., 139: 30-39.
CrossRefPubMedDirect Link - Abdollahi, M., M.Y. Moridani, O.I. Aruoma and S. Mostafalou, 2014. Oxidative stress in aging. Oxid. Med. Cell. Longev.
CrossRefDirect Link - Abe, A., T. Yamamoto, M. Isome, M. Ma and E. Yaoita et al., 1998. Thyroid hormone regulates expression of shaker-related potassium channel mRNA in rat heart. Biochem. Biophys. Res. Commun., 245: 226-230.
CrossRefPubMedDirect Link - Akao, M., A. Ohler, B. O'rourke and E. Marban, 2001. Mitochondrial ATP-sensitive potassium channels inhibit apoptosis induced by oxidative stress in cardiac cells. Circ. Res., 88: 1267-1275.
CrossRefPubMedDirect Link - Ardehali, H., 2004. Role of the mitochondrial ATP-sensitive K+ channels in cardioprotection. Acta Biochim. Pol., 51: 379-390.
PubMedDirect Link - Ardehali, H., B. O'Rourke and E. Marban, 2005. Cardioprotective role of the mitochondrial ATP-binding cassette protein 1. Circ. Res., 97: 740-742.
CrossRefPubMedDirect Link - Arsenijevic, D., H. Onuma, C. Pecqueur, S. Raimbault and B.S. Manning et al., 2000. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet., 26: 435-439.
CrossRefPubMedDirect Link - Arthur, J.R., 2000. The glutathione peroxidases. Cell. Mol. Life Sci., 57: 1825-1835.
CrossRefDirect Link - Bajgar, R., S. Seetharaman, A.J. Kowaltowski, K.D. Garlid and P. Paucek, 2001. Identification and properties of a novel intracellular (Mitochondrial) ATP-sensitive potassium channel in brain. J. Biol. Chem., 276: 33369-33374.
CrossRefDirect Link - Brand, M.D., K.M. Brindle, J.A. Buckingham, J.A. Harper, D.F. Rolfe and J.A. Stuart, 1999. The significance and mechanism of mitochondrial proton conductance. Int. J. Obes. Relat. Metab. Disord., 23: S4-S11.
PubMedDirect Link - Busija, D.W., T. Gaspar, F. Domoki, P.V. Katakam and F. Bari, 2008. Mitochondrial-mediated suppression of ROS production upon exposure of neurons to lethal stress: Mitochondrial targeted preconditioning. Adv. Drug Deliv. Rev., 60: 1471-1477.
CrossRefDirect Link - Cancherini, D.V., L.G. Trabuco, N.A. Reboucas and A.J. Kowaltowski, 2003. ATP-sensitive K+ channels in renal mitochondria. Am. J. Physiol. Renal. Physiol., 285: 1291-1296.
CrossRefPubMedDirect Link - Choudhury, S., G.B.N. Chainy and M.M. Mishro, 2003. Experimentally induced hypo- and hyper-thyroidism influence on the antioxidant defence system in adult rat testis. Andrologia, 35: 131-140.
CrossRefDirect Link - Cioffi, F., R. Senese, A. Lanni and F. Goglia, 2013. Thyroid hormones and mitochondria: with a brief look at derivatives and analogues. Mol. Cell. Endocrinol., 379: 51-61.
CrossRefPubMedDirect Link - Cohen, M.V., C.P. Baines and J.M. Downey, 2000. Ischemic preconditioning: From adenosine receptor to KATP channel. Annu. Rev. Physiol., 62: 79-109.
CrossRefDirect Link - Das, K. and G.B. Chainy, 2001. Modulation of rat liver mitochondrial antioxidant defence system by thyroid hormone. Biochem. Biophy. Acta, 1537: 1-13.
CrossRefPubMedDirect Link - Das, K. and G.B.N. Chainy, 2004. Thyroid hormone influences antioxidant defense system in adult rat brain. Neurochem. Res., 29: 1755-1766.
CrossRefDirect Link - De Castro, A.L., A.V. Tavares, C. Campos, R.O. Fernandes and R. Siqueira et al., 2014. Cardioprotective effects of thyroid hormones in a rat model of myocardial infarction are associated with oxidative stress reduction. Mol. Cell. Endocrinol., 391: 22-29.
CrossRefPubMedDirect Link - Debska, G., A. Kicinska, J. Skalska, A. Szewczyk, R. May, C.E. Elger and W.S. Kunz, 2002. Opening of potassium channels modulates mitochondrial function in rat skeletal muscle. Biochim. Biophys. Acta-Bioenerg., 1556: 97-105.
CrossRefPubMedDirect Link - Debska, G., R. May, A. Kicinska, A. Szewczyk, C.E. Elger and W.S. Kunz, 2001. Potassium channel openers depolarize hippocampal mitochondria. Brain Res., 892: 42-50.
CrossRefPubMedDirect Link - Droge, W., 2002. Free radicals in the physiological control of cell function. Phsyol. Rev., 82: 47-95.
CrossRefPubMedDirect Link - Dulloo, A.G. and S. Samec, 2001. Uncoupling proteins: Their roles in adaptive thermogenesis and substrate metabolism reconsidered. Br. J. Nutr., 86: 123-139.
CrossRefPubMedDirect Link - Forbes, R.A., C. Steenbergen and E. Murphy, 2001. Diazoxide-induced cardioprotection requires signaling through a redox-sensitive mechanism. Circ. Res., 88: 802-809.
CrossRefDirect Link - Forini, F., C. Kusmic, G. Nicolini, L. Mariani and R. Zucchi et al., 2014. Triiodothyronine prevents cardiac ischemia/reperfusion mitochondrial impairment and cell loss by regulating miR30a/p53 axis. Endocrinology, 155: 4581-4590.
CrossRefPubMedDirect Link - Forini, F., V. Lionetti, H. Ardehali, A. Pucci and F. Cecchetti et al., 2011. Early long-term L-T3 replacement rescues mitochondria and prevents ischemic cardiac remodelling in rats. J. Cell. Mol. Med., 15: 514-524.
CrossRefDirect Link - Forini, F., G. Nicolini and G. Iervasi, 2015. Mitochondria as key targets of cardioprotection in cardiac ischemic disease: Role of thyroid hormone triiodothyronine. Int. J. Mol. Sci., 16: 6312-6336.
CrossRefDirect Link - Gong, D.W., Y. He, M. Karas and M. Reitman, 1997. Uncoupling protein-3 is a mediator of thermogenesis regulated by thyroid hormone, β3-adrenergic agonists and leptin. J. Biol. Chem., 272: 24129-24132.
CrossRefDirect Link - Grover, G.J. and K.D. Garlid, 2000. ATP-Sensitive potassium channels: A review of their cardioprotective pharmacology. J. Mol. Cell. Cardiol., 32: 677-695.
CrossRefDirect Link - Halestrap, A.P., 1989. The regulation of the matrix volume of mammalian mitochondria in vivo and in vitro and its role in the control of mitochondrial metabolism. Biochim. Biophys. Acta (BBA)-Bioenergetics, 973: 355-382.
CrossRefDirect Link - Heinen, A., M. Aldakkak, D.F. Stowe, S.S. Rhodes, M.L. Riess, S.G. Varadarajan and A.K.S. Camara, 2007. Reverse electron flow-induced ROS production is attenuated by activation of mitochondrial Ca2+-sensitive K+ channels. Am. J. Physiol. Heart Circulatory Physiol., 293: 1400-1407.
CrossRefDirect Link - Hensely, K., K.A. Robinson, S.P. Gobbita, S. Salsman and R.A. Floyd, 2000. Reactive oxygen species, cell signaling and cell injury. Free Radic. Biol. Med., 28: 1456-1462.
CrossRefPubMedDirect Link - Holmuhamedov, E.L., L. Wang and A. Terzic, 1999. ATP-sensitive K+ channel openers prevent Ca2+ overload in rat cardiac mitochondria. J. Physiol., 519: 347-360.
CrossRefDirect Link - Inoue, I., H. Nagase, K. Kishi and T. Higuti, 1991. ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature, 352: 244-247.
CrossRefDirect Link - Jessell, T.M., 2000. Neuronal specification in the spinal cord: Inductive signals and transcriptional codes. Nat. Rev. Genet., 1: 20-29.
CrossRefDirect Link - Kloner, R.A., 1998. Ischemic preconditioning: The issues of refractoriness and tolerance. J. Am. Coll. Cardiol., 31: 1150-1151.
CrossRefPubMedDirect Link - Kowaltowski, A.J., S. Seetharaman, P. Paucek and K.D. Garlid, 2001. Bioenergetic consequences of opening the ATP-sensitive K+ channel of heart mitochondria. Am. J. Physiol. Heart Circulatory Physiol., 280: 649-657.
Direct Link - Kubo, Y., J.P. Adelman, D.E. Clapham, L.Y. Jan and A. Karschin et al., 2005. International union of pharmacology. LIV. Nomenclature and molecular relationships of inwardly rectifying potassium channels. Pharmacol. Rev., 57: 509-526.
CrossRefDirect Link - Kulawiak, B., A.P. Kudin, A. Szewczyk and W.S. Kunz, 2008. BK channel openers inhibit ROS production of isolated rat brain mitochondria. Exp. Neurol., 212: 543-547.
CrossRefDirect Link - Lanni, A., L. Beneduce, A. Lombardi, M. Moreno and O. Boss et al., 1999. Expression of uncoupling protein-3 and mitochondrial activity in the transition from hypothyroid to hyperthyroid state in rat skeletal muscle. FEBS Lett., 444: 250-254.
CrossRefDirect Link - Lanni, A., M.D. Felice, A. Lombardi, M. Moreno, C. Fleury, D. Ricquier and F. Goglia, 1997. Induction of UCP2 mRNA by thyroid hormones in rat heart. FEBS Lett., 418: 171-174.
CrossRefDirect Link - Lanni, A., M. Moreno, A. Lombardi and F. Goglia, 2003. Thyroid hormone and uncoupling proteins. FEBS Lett., 543: 5-10.
CrossRefDirect Link - Li, C. and R.M. Jackson, 2002. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am. J. Physiol. Cell Physiol., 282: C227-C241.
CrossRefPubMedDirect Link - Littleton, J.T. and B. Ganetzky, 2000. Ion channels and synaptic organization: Analysis of the Drosophila genome. Neuron, 26: 35-43.
CrossRefDirect Link - Liu, Y., T. Sato, B. O'Rourke and E. Marban, 1998. Mitochondrial ATP-dependent potassium channels: Novel effectors of cardioprotection? Circulation, 97: 2463-2469.
CrossRefDirect Link - Meyer, M., H.L. Pahl and P.A. Baeuerle, 1994. Regulation of the transcription factors NF-κB and AP-1 by redox changes. Chemico-Biol. Interact., 91: 91-100.
CrossRefDirect Link - Moher, D., A. Liberati, J. Tetzlaff and D.G. Altman, 2009. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. Br. Med. J., Vol. 339.
CrossRefDirect Link - Mourouzis, I., I. Giagourta, G. Galanopoulos, P. Mantzouratou and E. Kostakou et al., 2013. Thyroid hormone improves the mechanical performance of the post-infarcted diabetic myocardium: A response associated with up-regulation of Akt/mTOR and AMPK activation. Metabolism, 62: 1387-1393.
CrossRefDirect Link - Murata, M., M. Akao, B. O'Rourke and E. Marban, 2001. Mitochondrial ATP-sensitive potassium channels attenuate matrix Ca2+ overload during simulated ischemia and reperfusion: Possible mechanism of cardioprotection. Circulation Res., 89: 891-898.
CrossRefDirect Link - Murry, C.E., R.B. Jennings and K.A. Reimer, 1986. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation, 74: 1124-1136.
CrossRefDirect Link - Mutvei, A., S. Kuzela and B.D. Nelson, 1989. Control of mitochondrial transcription by thyroid hormone. Eur. J. Biochem., 180: 235-240.
CrossRefDirect Link - Naazeri, M., M. Rostamian and M. Hedayati, 2014. Impact of thyroid dysfunction on antioxidant capacity, super oxide dismutase and catalase activity. Zahedan J. Res. Med. Sci., 16: 51-54.
Direct Link - Nedergaard, J., D. Ricquier and L.P. Kozak, 2005. Uncoupling proteins: Current status and therapeutic prospects. EMBO Rep., 6: 917-921.
CrossRefDirect Link - Negre-Salvayre, A., C. Hirtz, G. Carrera, R. Cazenave and M. Troly et al., 1997. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J., 11: 809-815.
Direct Link - Nishiyama, A., F. Kambe, K. Kamiya, H. Seo and J. Toyama, 1998. Effects of thyroid status on expression of voltage-gated potassium channels in rat left ventricle. Cardiovasc. Res., 40: 343-351.
CrossRefDirect Link - Noma, A., 1983. ATP-regulated K+ channels in cardiac muscle. Nature, 14: 147-148.
CrossRefDirect Link - O'Rourke, B., 2000. Myocardial KATP channels in preconditioning. Circulation Res., 87: 845-855.
CrossRefDirect Link - O'Rourke, B., 2004. Evidence for mitochondrial K+ channels and their role in cardioprotection. Circulation Res., 94: 420-432.
CrossRefDirect Link - Ozcan, C., M. Bienengraeber, P.P. Dzeja and A. Terzic, 2002. Potassium channel openers protect cardiac mitochondria by attenuating oxidant stress at reoxygenation. Am. J. Physiol., 282: 531-539.
CrossRefDirect Link - Pantos, C. and D.V. Cokkinos, 2010. Thyroid hormone: An old drug to new indications. Vascular Pharmacol., 52: 1-101.
CrossRefDirect Link - Pantos, C., A. Dritsas, I. Mourouzis, A. Dimopoulos and G. Karatasakis et al., 2007. Thyroid hormone is a critical determinant of myocardial performance in patients with heart failure: Potential therapeutic implications. Eur. J. Endocrinol., 157: 515-520.
CrossRefDirect Link - Pantos, C., I. Mourouzis and D.V. Cokkinos, 2010. Thyroid hormone as a therapeutic option for treating ischaemic heart disease: From early reperfusion to late remodelling. Vascular Pharmacol., 52: 157-165.
CrossRefDirect Link - Pantos, C., I. Mourouzis and D.V. Cokkinos, 2011. New insights into the role of thyroid hormone in cardiac remodeling: Time to reconsider? Heart Fail Rev., 16: 79-96.
CrossRefDirect Link - Pantos, C., I. Mourouzis, G. Galanopoulos, M. Gavra, P. Perimenis, D. Spanou and D.V. Cokkinos, 2010. Thyroid hormone receptor α1 downregulation in postischemic heart failure progression: The potential role of tissue hypothyroidism. Horm. Metab. Res., 42: 718-724.
CrossRefPubMedDirect Link - Pantos, C., I. Mourouzis, T. Saranteas, V. Brozou, G. Galanopoulos, G. Kostopanagiotou and D.V. Cokkinos, 2011. Acute T3 treatment protects the heart against ischemia-reperfusion injury via TRα1 receptor. Mol. Cell. Biochem., 353: 235-241.
CrossRefDirect Link - Pantos, C., I. Mourouzis, T. Saranteas, G. Clave and H. Ligeret et al., 2009. Thyroid hormone improves postischaemic recovery of function while limiting apoptosis: A new therapeutic approach to support hemodynamics in the setting of ischaemia-reperfusion? Basic Res. Cardiol., 104: 69-77.
CrossRefDirect Link - Raha, S. and B.H. Robinson, 2000. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci., 25: 502-508.
CrossRefDirect Link - Ramsden, D.B., P.W.L. Ho, J.W.M. Ho, H.F. Liu and D.H.F. So et al., 2012. Human neuronal uncoupling proteins 4 and 5 (UCP4 and UCP5): structural properties, regulation and physiological role in protection against oxidative stress and mitochondrial dysfunction. Brain Behav., 2: 468-478.
CrossRefDirect Link - Ricquier, D. and F. Bouillaud, 2000. Mitochondrial uncoupling proteins: From mitochondria to the regulation of energy balance. J. Physiol., 529: 3-10.
CrossRefDirect Link - Rousset, S., M.C. Alves-Guerra, J. Mozo, B. Miroux, A.M. Casaard-Doulcier, F. Bouillaud and D. Ricquier, 2004. The biology of mitochondrial uncoupling proteins. Diabetes, 53: 130-135.
CrossRefDirect Link - Saeidnia, S. and M. Abdollahi, 2013. Toxicological and pharmacological concerns on oxidative stress and related diseases. Toxicol. Appl. Pharmacol., 273: 442-455.
CrossRefDirect Link - Sakaguchi, Y., G. Cui and L. Sen, 1996. Acute effects of thyroid hormone on inward rectifier potassium channel currents in guinea pig ventricular myocytes. Endocrinology, 137: 4744-4751.
CrossRefPubMedDirect Link - Salvemini, D. and S. Cuzzocrea, 2002. Oxidative stress in septic shock and disseminated intravascular coagulation. Free Radical Biol. Med., 33: 1173-1185.
CrossRefPubMedDirect Link - Short, K.R., J. Nygren, R. Barazzoni, J. Levine and K.S. Nair, 2001. T3 increases mitochondrial ATP production in oxidative muscle despite increased expression of UCP2 and-3. Am. J. Physiol.-Endocrinol. Metab., 280: E761-E769.
Direct Link - Silvestri, E., M. Moreno, A. Lombardi, M. Ragni and P. de Lange et al., 2005. Thyroid-hormone effects on putative biochemical pathways involved in UCP3 activation in rat skeletal muscle mitochondria. FEBS Lett., 579: 1639-1645.
CrossRefDirect Link - Skulachev, V.P., 1996. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q. Rev. Biophys., 29: 169-202.
CrossRefPubMedDirect Link - Skulachev, V.P., 1998. Uncoupling: New approaches to an old problem of bioenergetics. Biochim. Biophys. Acta: Bioenergetics, 1363: 100-124.
CrossRefDirect Link - Sreejith, P. and O.V. Oommen, 2006. Rapid regulatory effect of tri-iodothyronine (T3) on antioxidant enzyme activities in a fish Anabas testudineus (Bloch): Short-term in vivo and in vitro study. Indian J. Biochem. Biophys., 43: 119-122.
Direct Link - Suzuki, M., K. Kotake, K. Fujikura, N. Inagaki and T. Suzuki et al., 1997. Kir6.1: A possible subunit of ATP-sensitive K+ channels in mitochondria. Biochem. Biophys. Res. Commun., 241: 693-697.
CrossRefDirect Link - Suzuki, M., N. Sasaki, T. Miki, N. Sakamoto and Y. Ohmoto-Sekine et al., 2002. Role of sarcolemmal KATP channels in cardioprotection against ischemia/reperfusion injury in mice. J. Clin. Invest., 109: 509-516.
Direct Link - Szewczyk, A., W. Jarmuszkiewicz and W.S. Kunz, 2009. Mitochondrial potassium channels. IUBMB Life, 61: 134-143.
CrossRefDirect Link - Turrens, J.F., 2003. Mitochondrial formation of reactive oxygen species. J. Physiol., 552: 335-344.
CrossRefPubMedDirect Link - Hoek, T.L.V., L.B. Becker, Z. Shao, C. Li and P.T. Schumacker, 1998. Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J. Biol. Chem., 273: 18092-18098.
CrossRefDirect Link - Venditti, P. and S. di Meo, 2006. Thyroid hormone-induced oxidative stress. Cell. Mol. Life Sci., 63: 414-434.
CrossRefDirect Link - Vidal-Puig, A.J., D. Grujic, C.Y. Zhang, T. Hagen and O. Boss et al., 2000. Energy metabolism in uncoupling protein 3 gene knockout mice. J. Biol. Chem., 275: 16258-16266.
CrossRefDirect Link - Wrutniak-Cabello, C., F. Casas and G. Cabello, 2001. Thyroid hormone action in mitochondria. J. Mol. Endocrinol., 26: 67-77.
CrossRefDirect Link - Xu, M., Y. Wang, A. Ayub and M. Ashraf, 2001. Mitochondrial KATP channel activation reduces anoxic injury by restoring mitochondrial membrane potential. Am. J. Physiol.-Heart Circulatory Physiol., 281: H1295-H1303.
Direct Link - Yu, B.P. and H.Y. Chung, 2006. Adaptive mechanisms to oxidative stress during aging. Mech. Ageing Dev., 127: 436-443.
CrossRefDirect Link - Zhang, D.X., Y.F. Chen, W.B. Campbell, A.P. Zou, G.J. Gross and P.L. Li, 2001. Characteristics and superoxide-induced activation of reconstituted myocardial mitochondrial ATP-sensitive potassium channels. Circulation Res., 89: 1177-1183.
CrossRefPubMedDirect Link - Zhou, M., O. Tanaka, M. Sekiguchi, K. Sakabe and M. Anazi et al., 1999. Localization of the ATP-sensitive potassium channel subunit (Kir6.1/uKATP-1) in rat brain. Mol. Brain Res., 74: 15-25.
CrossRefPubMedDirect Link - Zhuo, M.L., Y. Huang, D.P. Liu and C.C. Liang, 2005. KATP channel: Relation with cell metabolism and role in the cardiovascular system. Int. J. Biochem. Cell Biol., 37: 751-764.
CrossRefPubMedDirect Link - McCord, J.M. and I. Fridovich, 1968. The reduction of cytochrome c by milk xantine oxidase. J. Biol. Chem., 243: 5753-5760.
PubMedDirect Link