
Wenjing Zhang
Department of Neurobiology, Key Laboratory for Neurodegenerative Disorder of the Ministry of Education, Beijing Institute of Brain Disorders, Capital Medical University, 10 Xitoutiao, Youanmen, Beijing 100069, People�s Republic of China
Haiting An
Department of Neurobiology, Key Laboratory for Neurodegenerative Disorder of the Ministry of Education, Beijing Institute of Brain Disorders, Capital Medical University, 10 Xitoutiao, Youanmen, Beijing 100069, People�s Republic of China
Feilong Zhang
Department of Neurobiology, Key Laboratory for Neurodegenerative Disorder of the Ministry of Education, Beijing Institute of Brain Disorders, Capital Medical University, 10 Xitoutiao, Youanmen, Beijing 100069, People�s Republic of China
Lin Dong
Department of Neurobiology, Key Laboratory for Neurodegenerative Disorder of the Ministry of Education, Beijing Institute of Brain Disorders, Capital Medical University, 10 Xitoutiao, Youanmen, Beijing 100069, People�s Republic of China
Qi Wang
Department of Neurobiology, Key Laboratory for Neurodegenerative Disorder of the Ministry of Education, Beijing Institute of Brain Disorders, Capital Medical University, 10 Xitoutiao, Youanmen, Beijing 100069, People�s Republic of China
Ruijun Su
Department of Neurobiology, Key Laboratory for Neurodegenerative Disorder of the Ministry of Education, Beijing Institute of Brain Disorders, Capital Medical University, 10 Xitoutiao, Youanmen, Beijing 100069, People�s Republic of China
Yanjing Qian
Department of Neurobiology, Key Laboratory for Neurodegenerative Disorder of the Ministry of Education, Beijing Institute of Brain Disorders, Capital Medical University, 10 Xitoutiao, Youanmen, Beijing 100069, People�s Republic of China
Xiaoli Gong
Department of Neurobiology, Key Laboratory for Neurodegenerative Disorder of the Ministry of Education, Beijing Institute of Brain Disorders, Capital Medical University, 10 Xitoutiao, Youanmen, Beijing 100069, People�s Republic of China
International Journal of Pharmacology
Year: 2015 | Volume: 11 | Issue: 1 | Page No.: 10-18







ABSTRACT
Parkinson’s Disease (PD) is a neurodegenerative disorder characterized by the progressive loss of dopaminergic (DA) neurons in the Substantia Nigra pars Compacta (SNc). There is no satisfactory therapeutic strategy for PD as yet. Here, triptolide (T10), a major active compound extracted from Tripterygium wilfordii Hook. f. (TWHF), a Traditional Chinese Medicine (TCM) used to treat autoimmune diseases, was found to significantly diminish the number of abnormal rotations induced by apomorphine in 6-hydroxydopamine (6-OHDA)-lesioned PD rat model and protected DA neurons in SNc from 6-OHDA toxicity. Triptolide treatment was found to significantly suppress the activation of microglia and reduce the levels of oxidation products of proteins and lipids in the striatonigral systems of the PD model rats. Results suggest that triptolide might be a suitable lead compound in drug discovery for PD therapy.
PDF Abstract XML References Citation
Received: October 20, 2014;
Accepted: December 10, 2014;
Published: January 13, 2015
How to cite this article
Wenjing Zhang, Haiting An, Feilong Zhang, Lin Dong, Qi Wang, Ruijun Su, Yanjing Qian and Xiaoli Gong, 2015. Triptolide Protects Dopaminergic Neurons from 6-OHDA Lesion in a Rat Model of Parkinson’s Disease. International Journal of Pharmacology, 11: 10-18.
DOI: 10.3923/ijp.2015.10.18
URL: https://scialert.net/abstract/?doi=ijp.2015.10.18
DOI: 10.3923/ijp.2015.10.18
URL: https://scialert.net/abstract/?doi=ijp.2015.10.18
INTRODUCTION
Parkinson’s Disease (PD), a common neurodegenerative disorder, which presents clinically as bradykinesia, muscular rigidity, resting tremor and postural instability, all of which are caused by the progressive loss of dopaminergic (DA) neurons in the Substantia Nigra pars Compacta (SNc) (Dauer and Przedborski, 2003; Meissner et al., 2011). Although the cause of idiopathic PD remains unknown, it is well-documented that increases in oxidative stress, inflammatory responses and mitochondrial dysfunction contribute to the cascade of events that cause the degeneration of DA neurons (Beal, 2005; Schapira and Jenner, 2011). Oxidative stress takes place through several mechanisms, such as depletion of antioxidant defenses, defects in mitochondrial electron transport, exposure to neurotoxins and oxidation of dopamine itself (Alam et al., 1997). 6-hydroxydopamine (6-OHDA) is one of the most common neurotoxins used in the establishment of PD models (Blum et al., 2001). This neurotoxin can enter the cell bodies and fibers of catecholaminergic neurons via the dopamine transporter (Deumens et al., 2002; Zhang et al., 2012). This can cause the formation of various oxidants and free radicals. In addition, 6-OHDA treatment reduces striatal glutathione (GSH) and Superoxide Dismutase (SOD) activity (Kumar et al., 1995; Schober, 2004). Together, the increasing oxidative stress due to 6-OHDA lesion triggers the progressive loss of DA neurons in the rat model of PD, which is therefore, an animal model considered appropriate for preclinical studies of PD therapy.
Currently, oral administration of L-dopa, the most commonly used medication for PD, is still the gold standard therapy which is effective for symptomatic relief during early stage of PD. Unfortunately, long-term administration of L-dopa (the precursor of dopamine), can cause side effects including dyskinesias and the "on-off" phenomenon (Del Sorbo and Albanese, 2008). There is currently a profound need for an efficient therapy with low toxicity for PD.
Triptolide, a diterpene triepoxide isolated from the medicinal plant Tripterygium wilfordii Hook. F. (TWHF), has been reported to exert multiple biological functions, including anti-inflammation, immunosuppression, neurotrophic and neuroprotective effects (Zheng et al., 2013). It acts as a potent antioxidant by inhibiting production of superoxide anion and NO, suppressing the expression of iNOS in murine peritoneal macrophage (Wu et al., 2006). It also increases the activity of endogenous antioxidant enzymes, e.g; SOD, catalase (CAT) and glutathione peroxidase (GSH-Px) in hepatic ischemia/reperfusion injury (Wu et al., 2011). Noticeably, pretreatment with triptolide were found to protect DA neurons in lipopolysaccharide-treated rats according to previous study (Zhou et al., 2005). However, the therapeutic effects of triptolide in symptomatic 6-OHDA-lesioned rat model of PD have not yet been reported.
In the present study, intraperitoneal administration of triptolide was found to improve behaviaural performance and prevent the loss of DA neurons in a rat model of PD induced by 6-OHDA. Subsequent observation of the antioxidant activity and microglial suppression of triptolide in 6-OHDA rats suggested that the underlying neuroprotective mechanism might involve reduced oxidative and inflammatory damage.
MATERIALS AND METHODS
Animals and treatments: Triptolide was purchased from Beijing Medicass Biotechnologies, Co., Ltd. Its molecular structure was identified by mass spectrometry and the white crystal was found to be 99% pure by High-Performance Liquid Chromatography (HPLC). To perform intraperitoneal injection, triptolide was resolved in normal saline containing 10% dimethyl sulfoxide.
Young adult male Sprague-Dawley rats weighing 220-240 g were supplied by the Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) and housed in a temperature-controlled (23±1°C) room under a standard 12 h light/dark cycle with ad libitum access to food and water in the laboratory animal center at Capital Medical University. All animal procedures were done following the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Animal Use and Care Committee of Capital Medical University. They were allowed to acclimate to the breeding environment for 7 days before experiments and all precautions were taken to minimize their suffering.
The rats were anesthetized with chloral hydrate (350 mg kg-1, i.p.) and then positioned in a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA, U.S.) with the tooth bar (set at -3.3 mm) and the blunt (45°) ear bars. Injections were made at the following stereotaxic coordinates: 1.0 mm anterior to bregma; 2.0 mm lateral to the midline and 5.5, 5.0 and 4.5 mm ventral to the dura. The 6-OHDA (Sigma, St. Louis, MO, U.S.) was diluted to 6 μg μL-1 in 0.9% saline (ascorbic acid, 0.2%) and injected at a flow rate of 1 μL min-1. After injection, the cannula remained in situ for additional 5 min and then withdrawn and a total volume of 3 μL was delivered to the striatum.
Rotational asymmetry analysis: After 5 weeks, unilateral 6-OHDA lesioned PD rat model was screened by subcutaneous apomorphine (APO) injection (0.5 mg kg-1, Sigma) induced rotational movement in automatic rotometer bowls (Columbus Instruments, U.S.). After rats were adapted to the testing environment, they were injected with APO at 0.5 mg kg-1. Measurements of rotational activity began 5 min after APO injection and then monitored for 30 min. The number of rotations per 30 min was calculated. During the test, the lights were turned off and the room was kept quiet. Counter-clockwise turns (ipsilateral to 6-OHDA injection) were counted as negative value and clockwise turns (contralateral to 6-OHDA injection) were counted as positive turns. The rats with >60 clockwise turns per 30 min were considered valid PD rats. This behavioral test was performed blindly.
Animal grouping: Rats were divided into four groups of nine to ten animals: (1) Sham group (striatal injection with saline) treated with intraperitoneal (i.p.) injection of saline once daily for 4 weeks, (2) model group (6-OHDA lesioned) treated with saline (i.p.) once daily for 4 weeks, (3) 6-OHDA-lesioned rats treated with triptolide (5 μg kg-1, i.p.) and (4) 6-OHDA-lesioned rats treated with triptolide (10 μg kg-1, i.p.) once daily for 4 weeks.
Immunohistochemical analysis: Rats were deeply anesthetized with chloral hydrate (350 mg kg-1, i.p.) and then transcardially perfused with 0.9% saline followed by 4% cold paraformaldehyde. Dissected brains were post-fixed in the same fixative for 4 h and cryoprotected in 30% sucrose solution for 3-5 days until they sank. The brains were frozen and arranged for frontal sectioning according to the rat brain atlas published by Paxinos and Watson. Brain coronal sections were cut of the striatum (30 μm in thickness, 0.26-1.6 mm posterior to bregma) or midbrain (50 μm in thickness, 4.80-6.30 mm posterior to bregma) with a cryostat (Leica, Germany) at -20°C and used for immunohistochemistry. Brain slices were incubated with primary Tyrosine Hydroxylase (TH) antibody (diluted 1:2000; Sigma, U.S.) or rabbit polyclonal anti-Iba1 (1:500; Wako, Osaka, Japan) for 24 h at 4°C and then detected using an ABC Elite kit (Vector laboratories, U.S.) with 3, 3-diaminobenzidine (DAB) to visualize immunoreactivity. TH immunoreactive (ir) neurons were stereologically counted only when they exhibited at least one neurite or had a visible nucleus using Stereo Investigator Software (MicroBrightField, Williston, VT, USA). The Optical Density (OD) of TH-ir fibers in the striatum was measured by quantifying optical segmentation of acquired 100× images. The average optic density value of Iba1-positive microglia in the SN of 20 consecutive sections of each brain was quantified using Image Pro Plus 6.0 software. The result was determined by the ratio of ipsilateral (left) side compared to the contralateral (right) side. Sections from three brains per group were coded and examined blindly.
Western blot analysis: Proteins were homogenized using the RIPA buffer (Beyotime, Beijing, China) containing protease inhibitor cocktail (1:100, Roche, Mannheim, Germany) and centrifuged at 12,000 rpm for 20 min at 4°C. Protein concentrations were measured by using a BCA kit (Thermo Fisher Scientific, Rockford, IL, USA). Equal protein samples were separated on 10% SDS polyacrylamide gels and then transferred to nitrocellulose membranes (Millipore) using a semidry blotting apparatus (Bio-Rad Laboratories, Hercules, CA). After blocking with 5% nonfat milk, membranes were labeled overnight at 4°C with a mouse antibody against TH (1:5000, Sigma) or β-actin (1:5000, Sigma). Next, membranes were washed with 0.5% Tween -20/PBS, incubated with an IRDye 800-labeled secondary antibody (1:20,000, Rockland Immunochemicals, Gilbertsville, PA, USA) and visualized with an Odyssey infrared imaging system (LI-COR instrument, Lincoln, NE, USA). The densitometry of TH were normalized to β-actin levels.
Measurement of oxidized proteins: Protein carbonyls in Ventral Mesencephalon (VM) and striatum were assayed by Western blot analysis, according to the manufacturer’s instruction (OxyBlot Protein Oxidation Detection Kit, Millipore, CA, U.S.). In brief, 20 μg protein samples from each group, 5 μL of 12% SDS and 10 μL of dinitrophenylhydrazine derivatizing agent were mixed and incubated at room temperature for 15 min. The reaction was terminated by adding 7.5 μL of neutralization buffer. Carbonyl derivatized samples were separated by 10% SDS-PAGE and then transferred to nitrocellulose membranes (Millipore). Membranes were blocked in a solution containing 1% BSA for 1 h at room temperature and labeled with a primary antibody (1:150) provided by the kit at 4 overnight. Next, the primary antibody-labeled membranes were treated with IRDyeTM 700-labelled secondary antibody (1:5000, Rockland) for 1 h at room temperature. The densitometric values of the oxyblot bands of 2, 4-dinitrophenyl (DNP)-modified proteins were visualized using the LI-COR Odyssey (LICOR, Lincoln, NE, U.S.) imaging system. β-actin served as a loading control.
ELISA: On the second day after assay of rotational behavior, three rats from each group were anesthetized and decapitated and their ventral mesencephalons and striatum were dissected quickly and stored in -80°C until analysis.
The ELISA was used to evaluate the contents of the antioxidant enzymes glutathione peroxidase (GSH-Px), SOD, CAT and malondialdehyde (MDA) in the brain samples in accordance with the user’s protocol included with the kit (Cusabio, Biotech Co., LTD, Newark, NJ, U.S.). Briefly, 100 mg tissue was rinsed and homogenized in 1 mL of 0.1 M phosphate buffer solution (pH 7.4). After two freeze-thaw cycles, the supernatants were assayed and the values were read at an absorption wavelength of 450 nm using a microplate reader (BioTEK, ELS800).
Statistical analysis: The data is presented as the Mean±Standard Error of the Mean (SEM). Statistical analysis was carried out using one-way ANOVA followed by Newman-Keuls post Test using Prism5.0 (GraphPad Software, San Diego, CA, U.S.). p<0.05 was considered statistically significant.
RESULTS
Triptolide significantly diminished the number of abnormal rotations in a rat model of PD: Firstly, the PD rat model was confirmed by apomorphine-induced rotation testing after 5 weeks of 6-OHDA unilateral lesion. Thereafter, the PD rats were subjected to triptolide (5 or 10 μg kg-1, i.p., once daily) for 4 weeks and rotational behavior induced by APO (0.5 mg kg-1, s.c.) was measured at 5, 6, 7 and 9 weeks after administration of 6-OHDA (Fig. 1a). As shown in Fig. 1b, the 6-OHDA rat underwent more than 100 rotations in 30 min. The rats in the sham group did not exhibit rotational behavior after APO challenge. There was no significant difference in rotational behavior response to APO among three groups: (1) 6-OHDA (model), (2) 6-OHDA+triptolide (5 μg kg-1) and (3) 6-OHDA + triptolide (10 μg kg-1) at 5 or 6 weeks after 6-OHDA lesion. Administration of triptolide at a dose of 10 μg kg-1 was found to alleviate the increase in the number of contralateral rotations of the PD rats at 7 and 9 weeks after 6-OHDA lesion (after triptolide treatment for 2 and 4 weeks, respectively). However no significant difference in rotational asymmetry from the model group was observed in the triptolide-treated PD rats (5 μg kg-1). These results demonstrate that triptolide (10 μg kg-1) can alleviate motor dysfunction in PD rats.
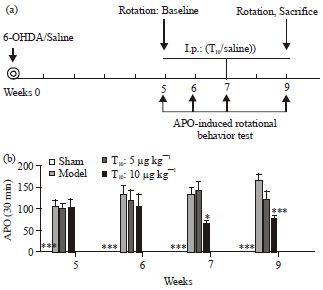 | |
| Fig. 1(a-b): | Triptolide attenuated rotational asymmetry in a rat model of PD, (a) Experimental structure, (b) Apomorphine-induced rotational asymmetry scores. T10, triptolide. Data is expressed as Mean±SEM One-way ANOVA followed by Newman-Keuls post test was applied. (n = 9-10 per group, *p<0.05, ***p<0.001 vs. model group within each group treated for different weeks) |
Triptolide treatment prevented the loss of TH-ir fibers in striatum and TH-ir neurons in SNc of 6-OHDA rat model of PD: The effect of triptolide on DA depletion was evaluated in 6-OHDA PD rats using the immunohistochemistry method with anti-TH antibody. Observations revealed that 6-OHDA exposure resulted in significantly fewer TH-immunoreactive (TH-ir) fibers in the left striatum (6-OHDA-injected striatum) relative to the right striatum of model rats than in those of sham rats. In contrast, extensive TH immunoreactivity was observed in lesioned striatum of triptolide (10 μg kg-1) rats at 9 weeks after 6-OHDA treatment (treatment with triptolide for 4 weeks) (Fig. 2a). Statistical analysis showed the optical density of TH-ir signals in lesioned striatum compared to the contralateral side of the triptolide (10 μg kg-1) treatment group to be significantly greater than in the model group (≈30%) (Fig. 2b). No significant difference in TH-ir fiber density was observed between the triptolide (5 μg kg-1) treatment group and the model group (Fig. 2a, b). TH-ir neurons were more readily detectable in the SNc of the sham group than in the model group (Fig. 2c). In the triptolide (10 μg kg-1) treatment group, the number of TH-ir cells in the ipsilateral SNc of the rats was ≈43% of the number in the contralateral SNc. This was significantly greater than that observed in the model group (≈27%) (Fig. 2c, d, p<0.05). There is no significant difference in TH-ir cell bodies between triptolide (5 μg kg-1) treatment groups and the model group (Fig. 2d). TH protein level of the four groups of rats was also determined by immunobloting and normalized to β-actin. The levels of TH protein in the both local striatum (≈14%) (Fig. 2e) and VM (≈46%) (Fig. 2g) in model group were markedly reduced compared to the sham group. However, the triptolide (10 μg kg-1) treatment prevented the decrease in TH levels in the both striatum (≈55%) (Fig. 2f, p<0.05) and local VM (≈80%) (Fig. 2h, p<0.001). While triptolide treatment in the dose of 5 μg kg-1 showed no significant changes compared to the model group (p>0.05).
Treatment with triptolide exerted anti-inflammatory effects on the brains of PD model rats: Microglial activation plays a critical role in the loss of DA neurons in SNc and it is also an important pathological feature of PD brains. To assess the effect of anti-microgliosis of triptolide in the 6-OHDA rat model, the expression of the microglial marker, Iba1 protein, was measured in brain sections from the sham group, model group and triptolide-treated groups (5 and 10 μg kg-1) using by immunohistochemical analysis.
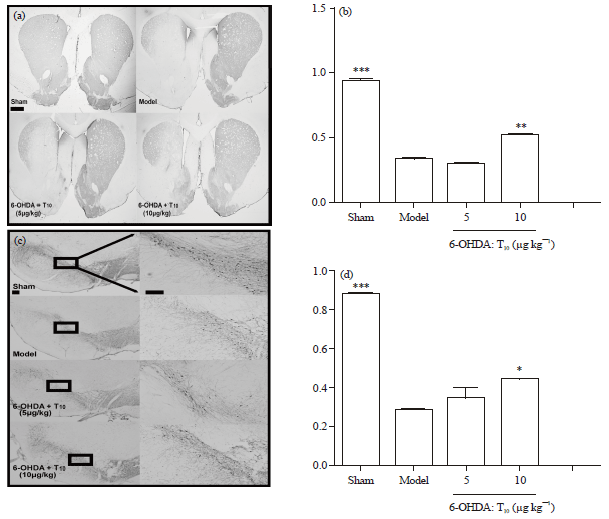 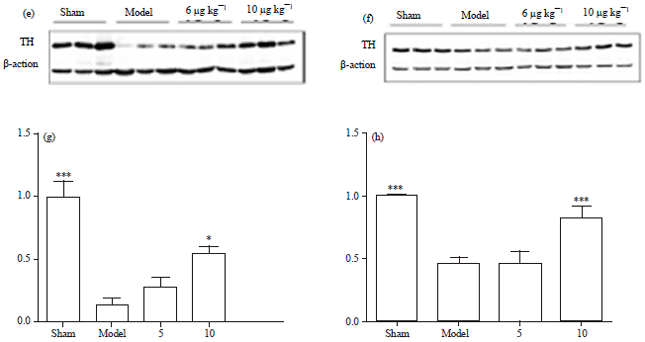 | |
| Fig. 2(a-h): | Triptolide prevented the loss of DA fibers in striatum and DA neurons in SNc of the PD rat model. TH immunocytochemistry was performed on coronal sections through, (a) Striatum and (c) SNc in representative rats from each group. (a) Scale bar = 1 mm, (c) Scale bars = 200 μm. (b) Relative density of TH-ir fibers in the injected striatum and the contralateral striatum. (d) Relative number of TH-ir neurons in the ipsilateral SNc compared to the contralateral SNc for the four different groups. The levels of TH protein in ipsilateral striatum (e) and VM (f) in representative rats were detected by western blot. The ratio of TH to β-actin in striatum (g) and VM (h) was calculated. T10, triptolide. Str, striatum. The data is shown as Mean±SEM. One-way ANOVA followed by Newman-Keuls post test was applied. (n = 3-4 per group, *p<0.05, **p<0.01, ***p<0.001 vs. model group) |
As shown in Fig. 3, microglia in the SNc of a rat from the sham group were characterized by lighter staining and fewer processes than in the SNc of the model group which exhibited activated microglia with thicker processes and more rounded cell bodies (Fig. 3a, b). The administration of triptolide (5 or 10 μg kg-1) for 4 weeks prevented the morphological changes observed in the microglia of the model brain (Fig. 3c, d). Quantification analysis of the Iba1-immunoreactivity (Fig. 3e) showed expression levels to be significantly higher in the SNc of model groups than in the sham groups while this increase was significantly reversed by triptolide treatment (10 μg kg-1). The data indicate that triptolide can suppress microglial activation in SNc of the PD rats.
Treatment with triptolide exerted anti-oxidative effects in the PD rat model: Because oxidative stress is an essential part of the phenotype of the 6-OHDA rat model of PD, oxidative levels of proteins and lipids were determined in the local VM and striatum of the four groups of rats. An OxyBlot kit and ELISA kit, respectively, were used to detect changes in the levels of carbonylated proteins, which may represent the extent of protein oxidation and MDA content, which is used to evaluate the degree of lipid oxidation in ipsilateral VM and striatum lysates. Here, 6-OHDA exposure produced significantly higher MDA content in both the VM and striatum than in the sham group. As expected, treatment with triptolide (10 μg kg-1) prevented the enhancement of MDA content in striatum and VM of the PD rats and triptolide at 5 μg kg-1 also exhibited a minimal effect in terms of MDA level only in striatum (Fig. 4a, b). Similarly, protein carbonyl formation in VM was triggered by 6-OHDA treatment and stabilized by triptolide (10 μg kg-1) administration, as shown in Fig. 4d. However, neither 6-OHDA lesion nor triptolide intervention influenced carbonyl levels of proteins in striatum. These data provide an indication that the lipid peroxidation is more readily detectable than protein oxidation after 9 weeks of 6-OHDA lesion and that oxidative stress could be prevented by administration of triptolide.
The ability of triptolide to modulate the levels of antioxidant enzymes was also analyzed. Kumar et al. (1995) reported that 6-OHDA injection in the striatum could reduce local levels of SOD and GSH-Px (Kumar et al., 1995). Here, results showed that 6-OHDA could decrease levels of SOD and GSH-Px in striatum to 60 and 64% sham-group levels, respectively. Administration of triptolide at 10 μg kg-1 prevented the decrease in the levels of GSH-Px and SOD in the striatum (Fig. 4e, f). Levels of another antioxidative enzyme, CAT, remained unaltered (Fig. 4g). Triptolide was here found to exert an antioxidative effect. This might be the main mechanism underlying its therapeutic action in PD rats.
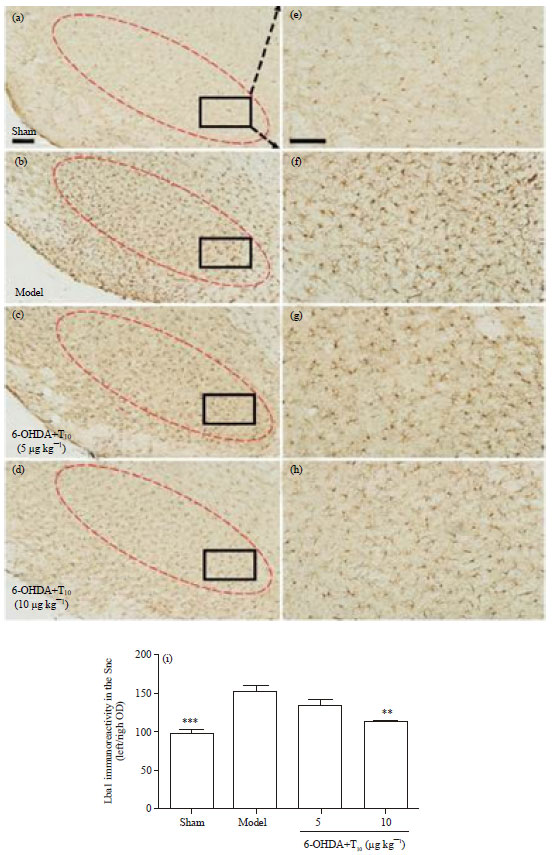 | |
| Fig. 3(a-i): | Administration of triptolide inhibited the activation of microglia in SNc of the PD rats, (a-d) Microglia in the SNc of the four groups of rats were detected using anti-Iba1 immunoreactivity staining. Scale bars = 200 μm, (e-h) High magnification of Iba1 staining in SNc bracketed in a-d. (i) Quantitative analysis of intensity of Iba1-immunoreactive images of the ipsilateral SNc (surrounded by dotted line in A-D) compared to the contralateral side from the four groups of rats. T10, triptolide. Data is expressed as Mean±SEM. One-way ANOVA followed by Newman-Keuls post test was applied (n = 3-4 per group, **p<0.01, ***p<0.001 vs. model group) |
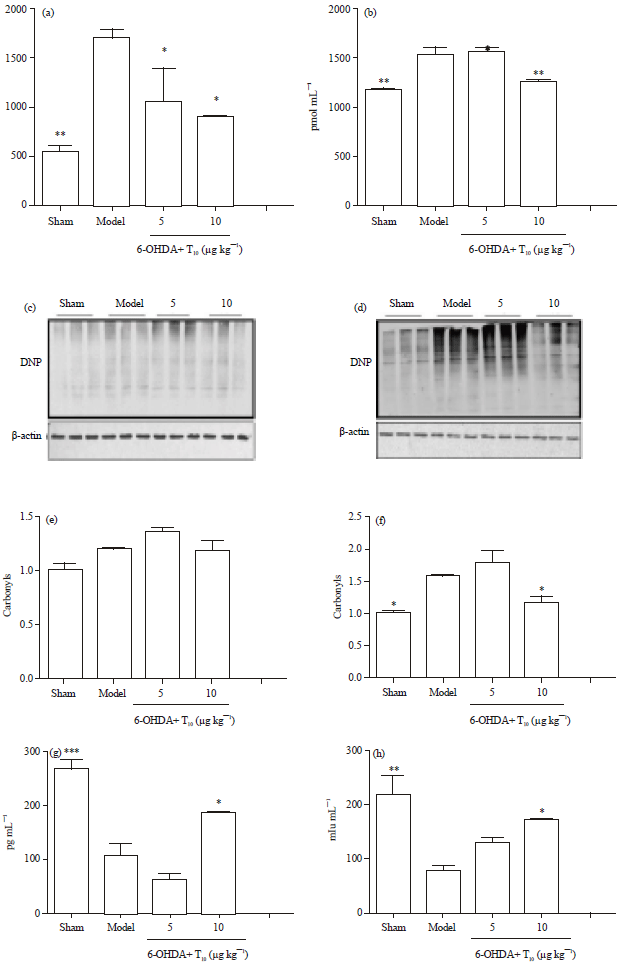 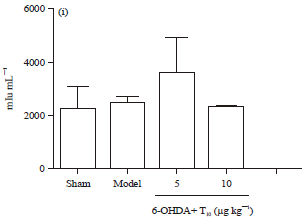 | |
| Fig. 4(a-i): | Treatment with triptolide exhibited anti-oxidative effects on the nigrostriatal system of the PD rats. MDA content in, (a) Striatum, (b) In ventral mesencephalons of four groups of rat. The protein samples extracted from, (c) Striatum, (d) ventral mesencephalons were treated with dinitrophenyl hydrazine and subjected to western blotting. (e-f) The levels of protein carbonyl formation were determined by quantifying the densitometry of positive bands of 2, 4-dinitrophenyl (DNP)-modified proteins. β-actin was used as loading control. (g-i) SOD, GSH-Px and CAT levels in the striatum of the four groups of rats were measured by ELISA and (i) T10, triptolide. Str, striatum. Data is expressed as Mean±SEM. One-way ANOVA followed by Newman-Keuls post test was applied. (n = 3 per group, *p<0.05, **p<0.01, ***p<0.001 vs. model group) |
DISCUSSION
We have reported that triptolide had preventive effect in Lipopolysaccharide (LPS)-induced rat model of PD at dose of 5 μg kg–1 (Zhou et al., 2005). The present study is the first to expand upon the investigation of triptolide in a symptomatic rat model of PD by unilateral striatal injection of 6-OHDA. This model is characterized by a gradual deterioration of the DA system over time and thus better resembles the natural DA degeneration that occurs in PD patients. This gives it a notable advantage in assessments of the anti-PD properties of new drugs (Dauer and Przedborski, 2003; Decressac et al., 2012). Here, triptolide alleviated the movement dysfunction of the 6-OHDA rats (Fig. 1a-b) similar to previous result, but with a higher effective dose than 5 μg kg-1 used in the LPS rat model of PD. The difference in dosage may be a cause of more deterioration and damage of DA system in the 6-OHDA rats than that in LPS rats.
Retrograde degeneration of DA neurons in the 6-OHDA rats is considered the pivotal cause of the abnormal rotations induced by APO. As expected, triptolide was found to prevent the loss of DA neurons in SNc and DA fibers in striatum as indicated by the TH protein immunocytochemistry of the PD rats (Fig. 2a-f).
6-OHDA lesion causes excessive microglial activation, which is believed to play a role in the development and progression of PD (Lee et al., 2009; Long-Smith et al., 2009). Here, results showed that triptolide could suppress microglial activation in SNc of the PD rats triggered by 6-OHDA (Fig. 3a-h), in accordance with other findings in LPS-lesioned PD rats (Zhou et al., 2005). The findings of this and other studies provide evidences of the anti-inflammatory of triptolide on a variety of tissues (Gao et al., 2008; Gong et al., 2008; Wang et al., 2008; Zhou et al., 2003). This suggests that triptolide alleviates the movement dysfunction and prevents loss of DA neurons of 6-OHDA rats due to extinguishing of the microglial activation in the process of PD pathology. However, the 6-OHDA-lesioned rats showed more generation of Reactive Oxygen Species (ROS) in cytosol of DA neurons in SNc than other models did. This caused the nigrostriatal pathway to degenerate (Dauer and Przedborski, 2003). In this regard, microglial activation may be secondary to the oxidative stress and may play an important role in exacerbating degeneration of the nigrostriatal system in 6-OHDA rats.
Here, this is found that lipids and protein oxidation being triggered by 6-OHDA lesion in the PD rats were prevented by triptolide administration (Fig. 4). The increase in protein and lipid peroxidation may be due to depletion of antioxidant defense capability caused by 6-OHDA (Blum et al., 2001; Chakraborty et al., 2013; Kumar et al., 1995). The present work mainly focuses on investigation of changes in levels of antioxidant enzymes, including SOD, GSH-Px and CAT and triptolide was found to stabilize the levels of these enzymes in striatum of 6-OHDA rats. This observation is supported by other studies performed in immune cells and hepatic disease (Wu et al., 2006, 2011). This suggests that triptolide might be an antioxidant in broad applications.
In conclusion, the present study shows that triptolide has both anti-inflammatory and anti-oxidative actions in the 6-OHDA-lestioned PD rats. Importantly, the ability of triptolide is observed to rapidly cross the blood brain barrier (data not shown). This could be a promising lead compound in drug discovery for PD therapy. However, the precise mechanism involved in this therapeutic effect remains to be delineated. For example, it is not clear whether the manner in which triptolide sustains the antioxidant defense suppresses microglial activation or whether this suppression sustains triptolide-induced antioxidant defense.
ACKNOWLEDGMENTS
This study was supported by the Chinese National Basic Research Program (2011CB504100), the National Natural Science Foundation of China (81030062, 31000476), the Important National Science and Technology Specific Project (2011ZX09102-003-01), the Projects of Beijing Educational Committee (IDHT20140514, KM201410025003, IJSHG201310025006).
REFERENCES
- Alam, Z.I., A. Jenner, S.E. Daniel, A.J. Lees and N. Cairns et al., 1997. Oxidative DNA damage in the parkinsonian brain: An apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J. Neurochem., 69: 1196-1203.
CrossRefPubMedDirect Link - Beal, M.F., 2005. Mitochondria take center stage in aging and neurodegeneration. Ann. Neurol., 58: 495-505.
CrossRefPubMedDirect Link - Blum, D., S. Torch, N. Lambeng, M.F. Nissou, A.L. Benabid, R. Sadoul and J.M. Verna, 2001. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: Contribution to the apoptotic theory in Parkinson's disease. Prog. Neurobiol., 65: 135-172.
CrossRefDirect Link - Chakraborty, S., J. Bornhorst, T.T. Nguyen and M. Aschner, 2013. Oxidative stress mechanisms underlying Parkinson's disease-associated neurodegeneration in C. elegans. Int. J. Mol. Sci., 14: 23103-23128.
CrossRefDirect Link - Dauer, W. and S. Przedborski, 2003. Parkinson's disease: Mechanisms and models. Neuron, 39: 889-909.
CrossRefPubMedDirect Link - Decressac, M., B. Mattsson and A. Bjorklund, 2012. Comparison of the behavioural and histological characteristics of the 6-OHDA and α-synuclein rat models of Parkinson's disease. Exp. Neurol., 235: 306-315.
CrossRefPubMedDirect Link - Del Sorbo, F. and A. Albanese, 2008. Levodopa-induced dyskinesias and their management. J. Neurol., 255: 32-41.
CrossRefPubMedDirect Link - Deumens, R., A. Blokland and J. Prickaerts, 2002. Modeling Parkinson's disease in rats: An evaluation of 6-OHDA lesions of the nigrostriatal pathway. Exp. Neurol., 175: 303-317.
CrossRefDirect Link - Gao, J.P., S. Sun, W.W. Li, Y.P. Chen and D.F. Cai, 2008. Triptolide protects against 1-methyl-4-phenyl pyridinium-induced dopaminergic neurotoxicity in rats: Implication for immunosuppressive therapy in Parkinson's disease. Neurosci. Bull., 24: 133-142.
CrossRefPubMedDirect Link - Gong, Y., B. Xue, J. Jiao, L. Jing and X. Wang, 2008. Triptolide inhibits COX-2 expression and PGE2 release by suppressing the activity of NF-κB and JNK in LPS-treated microglia. J. Neurochem., 107: 779-788.
CrossRefPubMedDirect Link - Kumar, R., A.K. Agarwal and P.K. Seth, 1995. Free radical-generated neurotoxicity of 6-hydroxydopamine. J. Neurochem., 64: 1703-1707.
CrossRefPubMedDirect Link - Lee, J.K., T. Tran and M.G. Tansey, 2009. Neuroinflammation in Parkinson's disease. J. Neuroimmune Pharmacol., 4: 419-429.
CrossRefPubMedDirect Link - Long-Smith, C.M., A.M. Sullivan and Y.M. Nolan, 2009. The influence of microglia on the pathogenesis of Parkinson's disease. Prog. Neurobiol., 89: 277-287.
CrossRefPubMedDirect Link - Meissner, W.G., M. Frasier, T. Gasser, C.G. Goetz and A. Lozano et al., 2011. Priorities in Parkinson's disease research. Nat. Rev. Drug Discovery, 10: 377-393.
CrossRefPubMedDirect Link - Schapira, A.H. and P. Jenner, 2011. Etiology and pathogenesis of Parkinson's disease. Mov. Disord., 26: 1049-1055.
CrossRefPubMedDirect Link - Schober, A., 2004. Classic toxin-induced animal models of Parkinson's disease: 6-OHDA and MPTP. Cell Tissue Res., 318: 215-224.
CrossRefPubMedDirect Link - Wang, Y., Y. Mei, D. Feng and L. Xu, 2008. Triptolide modulates T-cell inflammatory responses and ameliorates experimental autoimmune encephalomyelitis. J. Neurosci. Res., 86: 2441-2449.
CrossRefDirect Link - Wu, C., P. Wang, J. Rao, Z. Wang, C. Zhang, L. Lu and F. Zhang, 2011. Triptolide alleviates hepatic ischemia/reperfusion injury by attenuating oxidative stress and inhibiting NF-κB activity in mice. J. Surg. Res., 166: e205-e213.
CrossRefPubMedDirect Link - Wu, Y., J. Cui, X. Bao, S. Chan, D.O. Young, D. Liu and P. Shen, 2006. Triptolide attenuates oxidative stress, NF-κB activation and multiple cytokine gene expression in murine peritoneal macrophage. Int. J. Mol. Med., 17: 141-150.
PubMedDirect Link - Zhang, L.J., Y.Q. Xue, C. Yang, W.H. Yang and L. Chen et al., 2012. Human albumin prevents 6-hydroxydopamine-induced loss of tyrosine hydroxylase in in vitro and in vivo. PLoS ONE, Vol. 7.
CrossRefDirect Link - Zheng, Y., W.J. Zhang and X.M. Wang, 2013. Triptolide with potential medicinal value for diseases of the central nervous system. CNS Neurosci. Ther., 19: 76-82.
CrossRefPubMedDirect Link - Zhou, H.F., X.Y. Liu, D.B. Niu, F.Q. Li, Q.H. He and X.M. Wang, 2005. Triptolide protects dopaminergic neurons from inflammation-mediated damage induced by lipopolysaccharide intranigral injection. Neurobiol. Dis., 18: 441-449.
CrossRefPubMedDirect Link - Zhou, H.F., D.B. Niu, B. Xue, F.Q. Li and X.Y. Liu et al., 2003. Triptolide inhibits TNF-α, IL-1β and NO production in primary microglial cultures. Neuroreport, 14: 1091-1095.
CrossRefPubMedDirect Link