
Amany H. Mansour
Department of Clinical Pathology, Mansoura University, Egypt
Rokia Anwer
Department of Internal Medicine, Gastroenterology and Hepatology, Mansoura University, Egypt
Ahmad Darwish
Pediatric Hospital, Faculty of Medicine, Mansoura University, Egypt
Abdulrahman Fahmi Alshaik
Department of Emergency Hospital, Mansoura University, Egypt
Mohamed Mabed
Oncology Centre, Faculty of Medicine, Mansoura University, Egypt
International Journal of Pharmacology
Year: 2014 | Volume: 10 | Issue: 6 | Page No.: 315-321







ABSTRACT
Treatment of Acute Promyelocytic Leukemia (APL) with All–Trans Retinoic Acid (ATRA) represents a paradigm in targeted cancer therapy as these drugs cause clinical remission by affecting the stability of the fusion oncoprotein promyelocytic leukemia (PML)/Retinoic Acid Receptor Alpha (RARA). Previous studies have implicated in the main mechanism involved in therapy induced PML/RARA degradation. Investigated the effect of ATRA on the autophagy responses LC3–1, II and Beclin 1 proteins and if ATRA–induced upregulation of autophagy and Beclin 1 in the acute promyelocytic leukemia cells or not. Study included 22 subjects diagnosed as Acute Promyelocytic Leukemia (APL) (M3), also included 20 healthy subjects as a control selected to match the study. Peripheral blood and BM examination, Immunophenotyping, Conventional cytogenetic, Western blotting for LC3-I and LC3-II formation expression and Enzyme Link Immunosorbent Assay (ELISA) kit for Beclin-1 (BECN1). Both beclin-1 and LC3-11 were significantly increased in patient than control group before induction therapy. Proteins induced autophagy LC3-11 was increased after induction therapy than before in APL cells and that indicate autophagic degradation. The present study indicate that the ATRA-induced up regulation of autophagy protein and Beclin-1and LC3-1, 11 in the acute promyelocytic leukemia cells.
PDF Abstract XML References Citation
Received: June 22, 2014;
Accepted: August 07, 2014;
Published: October 01, 2014
How to cite this article
Amany H. Mansour, Rokia Anwer, Ahmad Darwish, Abdulrahman Fahmi Alshaik and Mohamed Mabed, 2014. All-Trans-Retinoid Acid (ATRA) Induced Upregulation of Autophagy Related Proteins (LC3–1, II and Beclin 1) in Acute Promyelocytic Leukemia. International Journal of Pharmacology, 10: 315-321.
DOI: 10.3923/ijp.2014.315.321
URL: https://scialert.net/abstract/?doi=ijp.2014.315.321
DOI: 10.3923/ijp.2014.315.321
URL: https://scialert.net/abstract/?doi=ijp.2014.315.321
INTRODUCTION
Leukemia, a malignant hematopoietic tumor, is a cancer of the blood or bone marrow which is characterized by the abnormal proliferation of white blood cells and is the 6th most common form of human cancer worldwide (Tallman and Altman, 2009).
Acute Myeloid Leukemia (AML) is characterized by an accumulation of abnormal hematopoietic progenitor cells which exhibit the morphology of a certain stage of myeloid differentiation in the bone marrow and the peripheral blood (Grignani et al., 1998). The blocked differentiation restrains progenitor cells from maturing and subsequently undergoing apoptosis and it may also enhance their self-renewal capabilities. Unlike normal hematopoietic cells that require cell extrinsic signals to maintain metabolism and survival, leukemia cells often express constitutively active oncogenic kinases that promote these processes independent of extrinsic growth factors. When cells receive insufficient growth signals or glucose metabolism decreases and the self-digestive process of autophagy is triggered to degrade bulk cytoplasm and organelles. Given its potential function in metabolism and cell survival, manipulation of autophagy may provide a critical means to eliminate certain type of leukemia cells (Bernardi and Pandolfi, 2007).
The clinical picture of APL is characterized by a rapidly increasing bleeding tendency which is due to pronounced coagulation disorders and severe thrombocytopenia. The microgranular variant (AML M3v) is a morphologically distinguishable special APL form which is mainly associated with increased leukocyte counts. The APL constitutes a hematological emergency which requires immediate diagnostic confirmation and specific therapeutic measures (Nasr et al., 2008). Diagnosis requires the APL-specific chromosomal translocation t (15,17) (q 22, q 21) and/or the fusion gene PML/RARA (Nasr et al., 2009).
The introduction of All-Trans-Retinoid Acid (ATRA) in APL therapy doubled the cure rate relative to chemotherapy alone. Remission rates of 80-90% and rates of long-term survival exceeding 75%, were reached with the combination of ATRA and anthracycline-containing chemotherapy (Chomienne et al., 1996). Yet, more than 10% of the patients treated in studies die in the course of induction therapy, mostly as a result of bleeding complications. All-Trans-Retinoic Acid (ATRA), a derivative of retinoic acid, reverses the differentiation block of promyelocytic blasts and induces the development of mature neutrophils, associated with a regression of the coagulation disorders within a few days. The extraordinarily high efficacy of ATRA is APL-specific (Lallemand-Breitenbach et al., 2008).
Autophagy is a ubiquitous process in which “bad” cytosolic molecules damaged organelles or invaded pathogens are sequestered within double-membrane vesicles called autophagosomes that deliver their contents to lysosomes for degradation and/or recycling of the resulting macromolecules. Indeed, autophagosomes were found to specifically target damaged mitochondria. Peroxisomes and protein aggregates that lead to DNA damage and genomic instability, thereby maintaining intracellular organelle and protein homeostasis (Tatham et al., 2008).
Autophagy may also provide nutrients through lysosome degradation of intracellular components of mitochondrial oxidation. Autophagy is more frequently compromised in solid cancer cells compared with their normal counterparts and acts as a protective mechanism in response to both extracellular and intracellular stress (Shen et al., 2004). Heterozygosity for the autophagy-essential gene. Beclin1 led to greatly increased rates of tumorigenesis, possibly because of genomic instability when autophagy is reduced (Bence et al., 2001).
The PML/RARA oncoprotein is known to be prone to aggregation, a feature that makes it a good substrate for autophagic degradation. The aggregation-prone characteristics of PML/RARA are further substantiated by showing that this protein chimera is considerably more effectively extracted by buffers containing the protein denaturation reagent UREA compared with buffers containing the detergents SDS or Triton X-100 (Pankiv et al., 2007). Its ability to form protein aggregates is possibly attributable to the PML moiety of the fusion autophagy and proteasome-dependent degradation may thus cooperate in therapy-induced clearance of the APL-associated oncoprotein. In line with this finding, proteolytic crosstalk has been reported to clearly exist between the autophagy and ubiquitin-proteasome systems (Hu et al., 2009).
Furthermore, proteolytic degradation of PML/RARA by caspases and lysosomal proteases has also been reported, suggesting the existence of multiple proteolytic pathways with a potential to target PML/RARA for degradation. However, because autophagy appears to become markedly induced in the presence of all-trans retinoic acid and arsenic trioxide, this degradation pathway may have a determinant role in therapy-induced PML/RARA clearance. Thus, both all-trans retinoic acid and arsenic trioxide induce clinical remission in APL patients by stimulating mTOR-dependent autophagy and causing concomitant autophagic degradation of the APL-associated oncoprotein PML/RARA (Nedelsky et al., 2008). In patients with the characteristic morphological picture of an APL and severe coagulation disorders, the onset of therapy with ATRA is justified even before genetic analysis confirms the diagnosis. ATRA is administered continuously until complete remission is obtained, at maximum up to a period of 90 days. ATRA monotherapy achieves a complete remission in 80-90 (%) of the patients with newly diagnosed APL. However, long-term remissions are rare under ATRA monotherapy (Rowe and Tallman, 1997). The aim of this study was to investigate the effect of ATRA on the autophagic responses LC3-1, II and Beclin 1 proteins.
METERIALS AND METHODS
This study was conducted in Mansoura Pediatric Hospital and Oncology center in Mansoura University hospital. It included that 22 patients (13 females and 9 males) diagnosed as Acute Promyelocytic Leukemia (APL) (M3). The study also included 20 healthy subjects (12 females and 8 males) as a control selected to match the study group in age and gender. The Ethics Committee of Mansoura, Faculty of Medicine approved the study protocol. Informed consent was taken before testing. All patients were subjected to the following.
Patient’s history and physical examination and full laboratory investigation include the following:
Peripheral blood and BM findings: In the AML cases, clinical studies of the PB, BM cellularity, according to WHO classification 2008 (Swerdlow and Campo, 2008).
Immunophenotyping: We reviewed all available data, including Peripheral Blood (PB) smears, Bone Marrow (BM) aspirates, cytochemical staining immunophenotyping of cells was performed with flow cytometry against CD3, CD41a, CD14, CD34, CD33, CD20, CD5, CD10, CD19, CD64, CD11c, CD13, CD117, CD56, CD2, CD7, HLA-DR, cytoplasmic CD22, cytoplasmic myeloperoxidase, cytoplasmic CD3 and cytoplasmic CD79a (coulter, San Jose, CA) (Dunphy et al., 2004).
Conventional cytogenetics: Chromosomal analyses were performed by examining short-term cultures of BM specimens according to standard conventional cytogenetic protocols. At least 20 cells in metaphase were analyzed in each case. Clonal abnormalities were classified according to the 2009 International System for Human Cytogenetic Nomenclature guideline (Golomb et al., 1976).
Fluorescence in situ hybridization: Fluorescence in situ Hybridization (FISH) analyses were performed to confirm translocation and the presence of t (15,17). These FISH analyses used pellets of cells remaining after conventional cytogenetic studies. Slides for FISH were prepared by using cells harvested for conventional cytogenetics and processing them for FISH according to the manufacturer's guidelines (Abbott V lysis, Des Plaines, IL, USA). (Gurrieri et al., 2004). Analyses were performed on cells in either interphase or metaphasedual-fusion probe (Abbott V lysis) was used to identify t (15;17) and its variants (Rowley et al., 1977).
Western blotting for LC3-I and LC3-II formation expression: First extracted in ice-cold Triton X-100 containing lysis buffer (50 mM NaCl, 10 mM Tris, 5 mM EDTA, 1% TritonX-100+protease and phosphatase inhibitor cocktails), the whole cell extract was harvested in lysis buffer. Cell lysates were then centrifuged (14000 rpm) for 10 min and the supernatants (soluble proteins) collected. The remaining protein pellets were washed with Phosphate Buffered Saline (PBS) before extraction with an SDS-containing buffer (2% SDS, 1mM DTT, 50mM Tris+protease and phosphatase inhibitor cocktails) to obtain SDS. The protein concentration was analyzed using a BCA assay (Thermo Fisher Scientific, Fremont, CA, USA). Protein extracts were loaded and resolved on 12% gels (Pierce, Rockford, IL) and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). After the whole cell extract had undergone western blotting, the SDS-PAGE PVDF membrane was blocked with antibodies against Rabbit anti-LC3 (Cell Signaling, 1:5000), LC3 (Novus Biologicals, Littleton, CO, USA), PARP-1/2 (H-250) and then incubated with secondary antibodies, anti-rabbit IgG antibody (Jackson Immuno-research, West Grove, PA, USA). Actin or α-tubulin (Sigma-Aldrich) was used as an internal control (Klionsky et al., 2008).
Enzyme-link immunosorbent assay (ELISA) kit for Beclin-1 (BECN1): Measurement the level of Beclin-1 (BECN1) by ELISA using USCNK life science InsUSA.Cut off 1.25 (ng L-1) (Sinha and Levine, 2008).
Statistical analysis: Results were collected, tabulated and statistically analyzed using statistical package version 16 (SPSS Inc., 2007). Continuous data was expressed as were eventually appropriate statistical tests. For normally distributed parameters group, comparison was made by use of paired and unpaired Student’s t-test. Mann-Whitney U-Test was used for non-normally distributed parameters. . In comparing 3 groups, one way ANOVA for statistical significance, were analyzed by the Mann-Whitney U-test (2 groups). Statistical significance was defined as p≤0.05 normally distributed data, presented as Mean±SD.
RESULTS
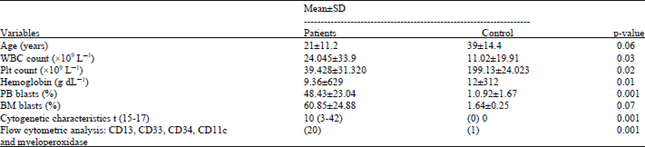
Peripheral blood analysis of our study revealed anemia (Hb 9.36±629 g dL-1) and leukocytosis (WBC 24.045±33.9x109 L-1). All AML patients also exhibited thrombocytopenia platelet count (39x109 L-1). The percentage of circulating blasts was variable and higher in AML patients (48%, p = 0.008). BM cellularity was nearly 100% and exhibited an increased number of blasts (60% in AML). Cytogenetic characteristics showed statistical significance (p = 00.1). Flow cytometric analysis showed a large population of myeloid blasts with the following immunophenotype; CD13, CD33, CD34, CD11c and myeloperoxidase (p = 0.001) (Table 1).
| Table 1: | Clinical and laboratory criteria in APL (M3) patients versus control before induction therapy |
 | |
 | |
| Fig. 1(a-b): | Autophagic measurement by changes in LC3 localization: LC3-1 and LC3-11 (a) After treatment and (b) Before treatment |
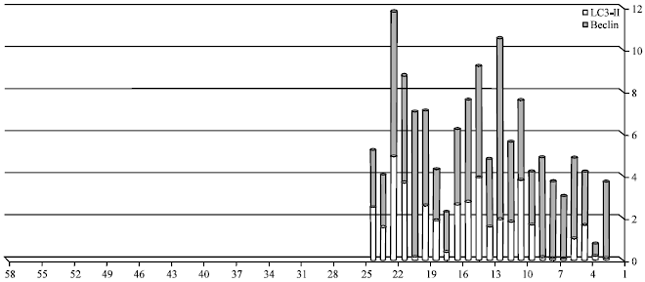 | |
| Fig. 2: | Level of Beclin-1 and LC3-11 in APL (M3) patients group after ATRA induction and M3 patients after induction the level of LC3-11 and beclin -1 (indicator of autophagic activity with autophagosome formation) |
| Table 2: | Levels of LC3-1,11 and beclin-1 in APL (M3) patients and control before treatment |
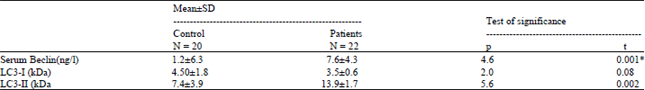 | |
There was significant increase in the level of Beclin-1 and LC3-II. On the other hand, non-significant difference with LC3-1 protein when compare with control before induction therapy (Table 2).
Autophagy can be measured by changes in LC3 localization; tracking the level of conversion of LC3-I to LC3-II provides an indicator of autophagic activity. In particular, the levels of LC3-II correlate with autophagosome formation due to its association with the autophagosome membrane. Western blotting (Fig. 1a, b) found that, LC3 is detected as two bands, cytosolic LC3-I and LC3-II which is bound to autophagosome membrane. This makes the molecular weight of LC3-II greater than LC3-I. However, due to its hydrophobicity, LC3-II migrates faster in SDS-PAGE and therefore displays a lower apparent molecular weight (Fig. 1a, b).
Table 3 and Fig. 2 show the levels of beclin-1 and LC3-11 in the total cases of AML M3 before treatment compared to their levels in the same cases after treatment.
| Table 3: | Beclin-1 and LC3-11 level in APL (M3) patients group before and after induction therapy |
 | |
There were significant increase in the levels of both markers in after treatment patients compared to before as following; beclin-1 (p = 0.001) , LC3-11 (p<0.059). The levels of the markers in the AML M3 patients after treatment were significantly higher than in pre-treatment patients (p = 0.059).
DISCUSSION
Autophagy is a critical modulator of cancer cell fate in virtue of its ability to regulate cell survival, differentiation and proliferation was investigated. In the present study, we investigated the link between autophagy, cell death and differentiation in the context of ATRA-induced granulocytic differentiation of the Acute Promyelocytic Leukemia (APL). We provide, here, the first evidence showing that Beclin 1 increased after ATRA induction therapy in APL M3 and this data is in accordance with Trocoli et al. (2011) who demonstrating that that Beclin- 1 is up regulated by ATRA in the myeloid leukemia and Beclin 1 displays a cytoprotective effect against numerous stressful situations. Such up regulations were also observed during the megakaryocytic differentiation of the chronic myelogenous leukemia, upporting the idea that autophagy and differentiation are interconnected processes (Bjorkoy et al., 2005).
Our study found an increased level of LC3-11after ATRA induction therapy (Table 3 and Fig. 2). Indicator of autophagic activity with autophagosome formation, consistent with the results of Nencioni et al. (2013) who reported that because ATRA and ATO-induced clinical remission is associated with degradation of PML/RARA, the ability of these drugs promote autophagy in NB4 cells. For these experiments we probed for the autophagy marker protein LC3 by Western blotting. Because LC3-II remains bound to autophagic membranes throughout the pathway, it is a useful marker to study autophagy and the LC3-II level is a good measure of autophagic activity. Suggesting that these drugs cause increased autophagic activity by using quantitative real-time polymerase chain reaction, he found that NB4 cells treated with ATRA or ATO had increased LC3 mRNA levels, indicating that these drugs also induce LC3 transcription. This newly synthesized LC3 is rapidly conjugated to LC3-II because there is no increase in the LC3-I levels (Lamark et al., 2009).
Interestingly, Wang et al. (2008) revealed that Beclin 1 exerts an anti-apoptotic effect by impeding the binding of the pro-apoptotic protein Bad to Bcl-XL, during the cell differentiation induced by vitamin D3. Similarly, Vazquez and Colombo (2010) demonstrated that the anti-apoptotic effect Coxiella burnetti (a gram negative bacterium) exerts in infected host cells is prevented by Beclin 1 depletion and by the expression of a Beclin 1 mutant with defective Bcl-2 binding, suggesting that the interaction of Beclin 1 and Bcl-2 may modulate apoptosis (Klionsky et al., 2008). These results support the idea that under some circumstances, the BH3-only domain of Beclin 1 may competitively disrupt the binding of pro-apoptotic proteins (e.g., Bad) to Bcl-2 or Bcl-XL, thus avoiding the induction of apoptosis. Paradoxically, the results of Ciechomska et al. (2009) showed that the binding of Beclin 1 to Bcl-2 does not modify the Bcl-2-mediated protection against apoptotic stimuli that initiates endoplasmic reticulum or mitochondria death signaling pathways. Thus, Beclin 1-regulated Bcl-2 anti-apoptotic function occurs only in certain circumstances (Youn et al., 2013).
These findings open the question as to whether or not the accumulation of Beclin 1 during ATRA-induced neutrophil/granulocyte differentiation of APL cells exerts an anti-apoptotic function by displacing the BH3-only pro-apoptotic proteins from their binding to Bcl-2 and Bcl-XL. Further studies are also required to elucidate the molecular mechanisms underlying Beclin 1 upregulation by ATRA. Chang et al. (2009) reported that in contrast to the effects of normal Atg13 levels, increased expression of Atg13 inhibits autophagosome expansion and recruitment of Atg8/LC3, potentially by decreasing the stability of Atg1 and facilitating its inhibitory phosphorylation by TOR. Atg1-Atg13 complexes, thus, function at multiple levels to mediate and adjust nutrient-dependent autophagic signaling. Interestingly,in addition, autophagy is involved in a novel form of programmed necrosis that occurs in neutrophils exposed to the inflammatory cytokine, GM-CSF (Zhang et al., 2009). Thus, the biological significance of Beclin 1 functions in normal neutrophil life span under both physiological and inflammatory conditions deserves to be investigated. In fact, to the best of our knowledge, there is no evidence in the literature showing that neutrophil function is abnormal in Atg-deficient mice (Liu et al., 2014).
It was found that inhibiting the ATRA-mediated Beclin 1 upregulation by a specific RNA directed against BECN1 did not impair ATRA-induced autophagy or the differentiation of APL cells, whereas it did affect the viability of mature APL cells. These findings suggest that Beclin- 1 had no crucial role in the induction of autophagy and differentiation in ATRA-treated APL cells. However, we cannot definitely rule out the role of Beclin 1 in these processes since extinction of Beclin 1 by siRNA was not complete in our experiments and this may be insufficient to prevent autophagy and differentiation induced by ATRA (Maycotte and Thorburn, 2011).
CONCLUSION
It was concluded that the ATRA-induced upregulation of autophagy protein and Beclin-1 and LC3-1, 11 in the acute promyelocytic leukemia cells. Most importantly, because PML/RARA proteolysis appears to be critical for APL remission, this data may provide new options for therapy-resistant patients because drugs known to induce autophagy already are used in the clinic.
REFERENCES
- Tallman, M.S. and J.K. Altman, 2009. How I treat acute promyelocytic leukemia. Blood, 114: 5126-5135.
CrossRefPubMedDirect Link - Grignani, F., S. de Matteis, C. Nervi, L. Tomassoni and V. Gelmetti et al., 1998. Fusion proteins of the retinoic acid receptor-α recruit histone deacetylase in promyelocytic leukaemia. Nature, 391: 815-818.
CrossRefDirect Link - Bernardi, R. and P.P. Pandolfi, 2007. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol., 8: 1006-1016.
CrossRefDirect Link - Nasr, R., M.C. Guillemin, O. Ferhi, H. Soilihi and L. Peres et al., 2008. Eradication of acute promyelocytic leukemia-initiating cells through PML-RARA degradation. Nat. Med., 14: 1333-1342.
CrossRef - Lallemand-Breitenbach, V., M. Jeanne, S. Benhenda, R. Nasr and M. Lei et al., 2008. Arsenic degrades PML or PML-RARα through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat. Cell Biol., 10: 547-555.
CrossRef - Tatham, M.H., M.C. Geoffroy, L. Shen, A. Plechanovova and N. Hattersley et al., 2008. RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat. Cell Biol., 10: 538-546.
CrossRef - Shen, Z.X., Z.Z. Shi, J. Fang, B.W. Gu and J.M. Li et al., 2004. All-trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA., 101: 5328-5335.
CrossRef - Bence, N.F., R.M. Sampat and R.R. Kopito, 2001. Impairment of the ubiquitin-proteasome system by protein aggregation. Science, 292: 1552-1555.
CrossRefPubMedDirect Link - Pankiv, S., T.H. Clausen, T. Lamark, A. Brech and J.A. Bruun et al., 2007. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem., 282: 24131-24145.
CrossRefDirect Link - Hu, J., Y.F. Liu, C.F. Wu, F. Xu and Z.X. Shen et al., 2009. Long-term efficacy and safety of all-trans retinoic acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA., 106: 3342-3347.
CrossRef - Nedelsky, N.B., P.K. Todd and J.P. Taylor, 2008. Autophagy and the ubiquitin-proteasome system: Collaborators in neuroprotection. Biochimica Biophysica Acta (BBA)-Mol. Basis Dis., 1782: 691-699.
CrossRef - Chomienne, C., P. Fenaux and L. Degos, 1996. Retinoid differentiation therapy in promyelocytic leukemia. FASEB J., 10: 1025-1030.
Direct Link - Dunphy, C.H., S.O. Orton and J. Mantell, 2004. Relative contributions of enzyme cytochemistry and flow cytometric immunophenotyping to the evaluation of acute myeloid leukemias with a monocytic component and of flow cytometric immunophenotyping to the evaluation of absolute monocytoses. Am. J. Clin. Pathol., 122: 865-874.
CrossRefDirect Link - Golomb, H.M., J. Rowley, J. Vardiman, J. Baron, G. Locker and S. Krasnow, 1976. Partial deletion of long arm of chromosome 17: A specific abnormality in acute promyelocytic leukemia? Arch. Internal Med., 136: 825-828.
CrossRef - Gurrieri, C., K. Nafa, T. Merghoub, R. Bernardi and P. Capodieci et al., 2004. Mutations of the PML tumor suppressor gene in acute promyelocytic leukemia. Blood, 103: 2358-2362.
CrossRef - Klionsky, D.J., H. Abeliovich, P. Agostinis, D.K. Agrawal and G. Aliev et al., 2008. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy, 4: 151-175.
PubMed - Sinha, S. and B. Levine, 2008. The autophagy effector Beclin 1: A novel BH3-only protein. Oncogene, 27: S137-S148.
CrossRefDirect Link - Bjorkoy, G., T. Lamark, A. Brech, H. Outzen and M. Perander et al., 2005. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol., 171: 603-614.
PubMedDirect Link - Nencioni, A., M. Cea, F. Montecucco, V.D. Longo and F. Patrone et al., 2013. Autophagy in blood cancers: Biological role and therapeutic implications. Haematologica, 98: 1335-1343.
CrossRefDirect Link - Lamark, T., V. Kirkin, I. Dikic and T. Johansen, 2009. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle, 8: 1986-1990.
Direct Link - Wang, J., H. Lian, Y. Zhao, M.A. Kauss and S. Spindel, 2008. Vitamin D3 induces autophagy of human myeloid leukemia cells. J. Biol. Chem., 283: 25596-25605.
CrossRefPubMedDirect Link - Vazquez, C.L. and M.I. Colombo, 2010. Coxiella burnetii modulates Beclin 1 and Bcl-2, preventing host cell apoptosis to generate a persistent bacterial infection. Cell Death Differ., 17: 421-438.
CrossRefDirect Link - Ciechomska, I.A., G.C. Goemans, J.N. Skepper and A.M. Tolkovsky, 2009. Bcl-2 complexed with Beclin-1 maintains full anti-apoptotic function. Oncogene, 28: 2128-2141.
CrossRef - Youn, H., E.J. Kim and S.J. Um, 2013. Zyxin cooperates with PTOV1 to confer retinoic acid resistance by repressing RAR activity. Cancer Lett., 331: 192-199.
CrossRef - Chang, Y.Y., G. Juhasz, P. Goraksha-Hicks, A.M. Arsham, D.R. Mallin, L.K. Muller and T.P. Neufeld, 2009. Nutrient-dependent regulation of autophagy through the target of rapamycin pathway. Biochem. Soc. Trans., 37: 232-236.
Direct Link - Zhang, Y., S. Goldman, R. Baerga, Y. Zhao, M. Komatsu and S. Jin, 2009. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc. Nat. Acad. Sci., 106: 19860-19865.
CrossRef - Liu, Z., T. Li, K. Jiang, Q. Huang, Y. Chen and F. Qian, 2014. Induction of chemoresistance by all-trans retinoic acid via a noncanonical signaling in multiple myeloma cells. PLoS One, Vol. 9.
CrossRef - Maycotte, P. and A. Thorburn, 2011. Autophagy and cancer therapy. Cancer Biol. Ther., 11: 127-137.
Direct Link