
Y.H. Siddique
Human Genetics and Toxicology Laboratory, Section of Genetics, Department of Zoology, Faculty of Life Sciences, Aligarh Muslim University, Aligarh (UP)-202002, India
M. Afzal
Human Genetics and Toxicology Laboratory, Section of Genetics, Department of Zoology, Faculty of Life Sciences, Aligarh Muslim University, Aligarh (UP)-202002, India
International Journal of Pharmacology
Year: 2008 | Volume: 4 | Issue: 6 | Page No.: 410-430







ABSTRACT
The present review gives the details of the genotoxic studies carried out till date for some selected synthetic progestins. Mutagenicity is defined as a permanent change in content or structure of the genetic material of an organism. A mutagenic hazard can be manifested as a heritable change resulting from germ-line mutations and/or somatic mutations leading to cancer or other chronic degenerative processes such as aging. Reactive Oxygen Species (ROS) generated through normal metabolic processes or from toxic products, can lead to a state of oxidative stress that contributes to the pathogenesis of a number of human disease by damaging lipids, protein and DNA. Oral contraceptives have been used since the early 1960s and are now used by about 90 million women world wide. The pill is given as a combination of an estrogen and a progestogen. The estrogen component of combined oral contraceptives is either ethinylestradiol or mestranol and the progestogens used are cyproterone acetate, desogestrol, ethynodiol diacetate, levonorgestrel, lynestrenol, megestrol acetate, norethisterone, norethisterone acetate, norethynodrel, norgestimate and norgestrel. Little is known about the long term health risks and potential protective effects of these individual components. Synthetic progestins induced the genotoxic damage and also various types of cancers, both singly as well as in combination with estrogens. Various synthetic progestins have been tested for their genotoxic effects in different experimental models, using different genotoxic end points. Ethynodioldiacetate, norethynodrel, norgestrel, lynestrenol and medroxyprogesterone acetate were found to be genotoxic only in the presence of metabolic activation supplemented with NADP. Megestrol acetate, cyproterone acetate and chlormadinone acetate were found to be genotoxic in the absence of metabolic activation. On the basis of reports available it is suggested that the progestins in which double bond between carbon-6 and carbon-7 is present, they undergo nucleophilic reaction and generates free radical in the system to show the genotoxic effects and the progestins in which double bond between carbon-6 and carbon-7 is absent, they need metabolic activation like estrogens, such as estradiol-17β and ethinylestradiol to show the genotoxic effects.
PDF Abstract XML References Citation
How to cite this article
Y.H. Siddique and M. Afzal, 2008. A Review on the Genotoxic Effects of Some Synthetic Progestins. International Journal of Pharmacology, 4: 410-430.
DOI: 10.3923/ijp.2008.410.430
URL: https://scialert.net/abstract/?doi=ijp.2008.410.430
DOI: 10.3923/ijp.2008.410.430
URL: https://scialert.net/abstract/?doi=ijp.2008.410.430
INTRODUCTION
Steroid hormones are the members of lipid compounds. They are mainly secreted by adrenal cortex of mammals, testis in male, ovary in the female and the placenta of mammals. Steroids consist of a tetracyclic nucleus, which is named cyclopentanoperhydro-phenanthrene (Fig. 1).
The phenanthrene portion of the nucleus is comprised of rings A, B and C, while D is the cyclopentane portion. Steroid hormones are responsible for a number of physiological and pharmacological effects in humans and other mammals. There are four basic classes of steroid hormones (Gorbman et al., 1983).
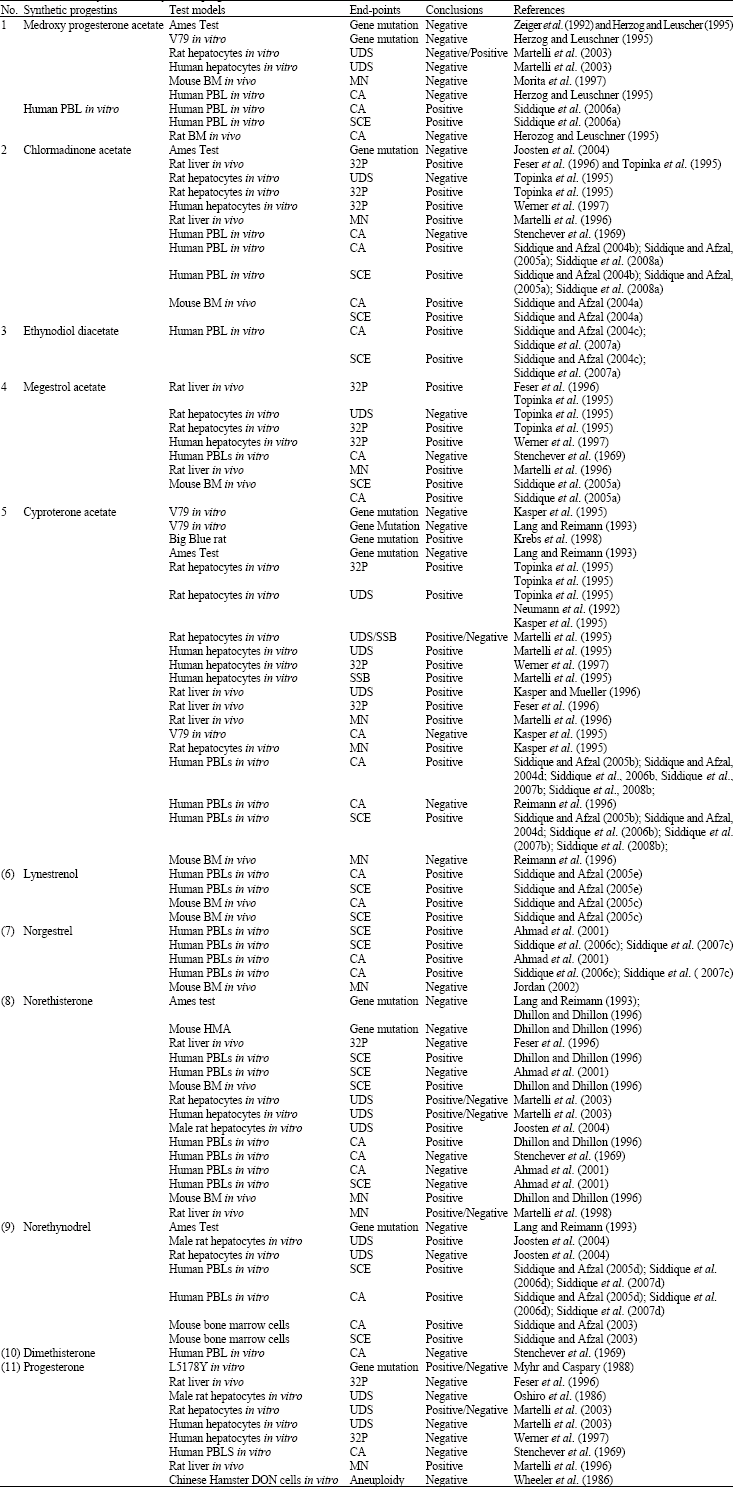
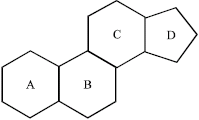 | |
| Fig. 1: | Steroid nucleus |
Adenocorticoids: Adrenocorticoids are grouped into the (i) Glucocorticoids: Cortisol, corticosterone and cortisone. They facilitate the formation of carbohydrates from non-carbohydrate sources (gluconeogenesis) and (ii).
Mineralocorticoids: They have ability to affect water and electrolyte metabolism (favoring retention of Na+ and excretion of K+), e.g., Aldosterone.
Estrogens: Estrogens have the ability (secreted by the ovarian follicle) to stimulate female secondary sex characteristics and to help maintain the female reproductive tract e.g., estrone, estradiol and estriol.
Progestins: They stimulate the uterus and maintain uterine development during pregnancy (secreted by the corpus luteum of the ovary) e.g., Progesterone, 17α-hydroxyprogesterone.
Androgens: They stimulate male characteristics and maintain male sex accessory glands and ducts (secreted by testis) e.g., testosterone, 5 α-dihydrotestosterone and androstenedione. The usefulness of above classification scheme is limited to some mammalian species, because glucocorticoids stimulates aspects of female reproduction on in bony fishes. Aldosterone is usually classified as a mineralocorticoid, but it is a potent glucocorticoid in some animals.
Structure and nomenclature of steroids: The basic nucleus of steroids has 17 carbon atoms as shown in the Fig. 2.
The nucleus has six asymmetric carbon atoms, shown with asterisk in the Fig. 2. The nucleus is a flat structure lying in the plane of the page. If the hydrogen projects towards the observer, it is said to be in the cis or-β position (solid line) and if the hydrogen project away from the observer, it is said to be in the trans or α-position (broken lines) as shown below:
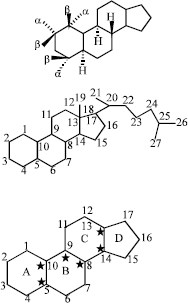 | |
| Fig. 2: | Structure of steroids |
Additional carbons can be attached to the 17-carbon nucleus to form other classes of steroid compounds. If a methyl atom is attached to carbon 13, a new compound known as estrone (C-18) is formed. If a second methyl group is added to carbon 10, androstane (C-19) is formed.
Metabolism of steroids: Ovary, testis, adrenal gland and placenta have the ability to produce steroidal hormones, which is favoured by the trophic hormones such as andrenocorticotrophic Hormone (ACTH), Luetinizing Hormone (LH), Follicle Stimulating Hormone (FSH) and Progesterone Releasing Hormone (PRL) and its receptor in the target tissues. It is also possible that one steroid secreting organ can take up steroid originally secreted by another organ and modified its activity. For example, mammalian adrenal cortex can take up progesterone secreted by the ovary and modifies it to corticoids. Chemical modifications may also occur in the brain, skin, salivary glands and other tissues. Steroid hormones are synthesized from a C27 precursor steroid, cholesterol. There are multiple sources of this steroid hormone precursor: de novo synthesis within the endocrine organ and uptake from the circulating pool that is maintained by the liver or supplied in the diet. In the blood cholesterol circulates in the form of cholesterol esters bound to Low and High Density Lipoprotein (LDL and HDL). For the synthesis of the steroid hormone the breaking off of the six carbon side chain of the cholesterol occurs in mitochondria (Gorbman et al., 1983).
The inner membrane of the mitochondria contains an oxygenases known as cytochrome P450, which breaks the side chain of the cholesterol and results in the formation of pregnenolone (C21). Pregnenolone is further transported to endoplasmic reticulum, where it is either oxidized to form progesterone or hydroxylated to form 17-hydroxypregnenolone. Further with the help of hydroxylating enzyme, desmolase and various cofactors triphosphopyridine nucleotide (TPNH) and molecular oxygen give rise to various steroid hormones (Fig. 3). The catabolism of steroid occurs in both steroidogenic and peripheral tissues. The major site of catabolism is the liver although the kidney and other tissues may catabolize circulating steroids to some extent. The major catabolic enzymes are involved in reductive reactions, particularly in the ring A and carbons 3 and 20. They are reduced to more water soluble and in active steroid sulfates and glucoronides that are eliminated through the urine or through the bile fluid, with ultimate excretion in the feaces (Gorbman et al., 1983).
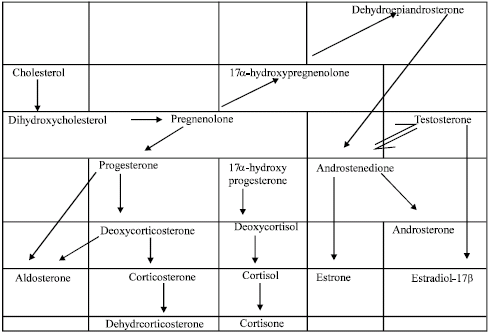 | |
| Fig. 3: | Biogenesis of steroid hormones |
Action of Birth Control Pills (BCPs): Normally the pituitary glands produces two hormones called Follicle Stimulating Hormone (FSH) and Leutenizing Hormone (LH). These hormones serve to stimulate the ovary to produce an egg at each menstrual cycle (to ovulate). The ovary is also the production site for the woman`s two central female hormones estradiol and progesterone, (Progestin) (Elstein et al., 1976). Birth Control Pills (BCPs) are a combination of synthetic estrogen and progestin. BCPs fool the pituitary gland so that it produces less FSH and LH. By reducing the FSH and LH required for ovulation, birth control pills suppress, but do not eliminate ovulation. BPCs may have two main effects (Wolf et al., 1979; Chang and Hunt, 1970): (i) They thin the inner lining of the uterus (called the endometrium) depleting it of glycogen and blood supply, (ii) BCPs may thicken the cervical mucus, making it more difficult for the sperm to travel up through the cervix. Intergrins are a group of adhesion molecules that have been implicated as playing an important role in fertilization and implantation. According to Somkuti et al. (1996) there are several types of integrins and it is believed that the endometrium is most receptive to implantation when it expresses certain types of integrins. BPCs change the type of integrins that the endometrial lining produces and makes the implantation difficult of the pre-born child (Somkuti et al., 1996).
Genotoxic damage and cancer risks of oral contraceptives: The type of oral contraceptives prescribed differs between countries and both the type of oral contraceptive and the doses of estrogens and progestogens have changed between and within countries overtime (Joosten et al., 2004). Chromosomal abnormalities and sister chromatid exchanges have been reported in peripheral blood lymphocytes of women taking oral contraceptives (Carr, 1967, 1970; Goh, 1967; Murthy and Prema, 1979). There is sufficient data which indicates that the prolonged use of the synthetic progestins can cause cancer among animals and humans (Rudali, 1975; El-Etreby and Gräf, 1979; Misdorp, 1991; Deml et al., 1993; Martelli et al., 1996; Heinemann et al., 1997; Maier and Herman, 2001).
SYNTHETIC PROGESTINS
Synthetic progestins have a wide spread use in medicine but their side effects are often debatable. Due to the genotoxic hazards and carcinogenic risks of synthetic progestins it is important to review the studies carried out till date. The present review is limited to some commonly use synthetic progestins in oral contraceptives formulations. Progestins were first isolated in 1993 and progesterone itself was synthesized in the 1940s. There are different classes of progestins, such as progesterone, retroprogesterone, progesterone derivative, 17α-hydroxy progesterone derivatives (pregnanes), 17α-hydroxy nor progesterone derivatives (non-pregnanes), 19-norprogesterone derivatives (nor pregnanes), 19-nortestosterone derivatives (estranes), 19-nortestosterone derivatives (gonanes) and spirolactone derivative. Schindler et al. (2003) has classified progestins in nine groups (Table 1) and the details for the genotoxic studies carried out till date is given in Table 2.
| Table 1: | Types of Progestins |
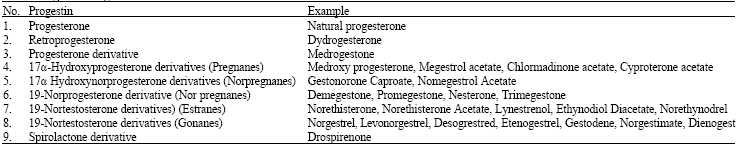 | |
| Table 2: | Genotoxicity studies for synthetic progestins carried out till date |
 | |
Medroxy progesterone acetate (CAS: 71-58-9)
(16)-17-(Acetyloxy)-6-methylpregn-4-ene-3,20-dione)
Colour: White or off-white
Odour: Odourless
Solubility: Medroxyprogesterone acetate is practically insoluble in water, sparingly soluble in alcohol and in methyl alcohol, slightly soluble in ether freely soluble in chloroform, acetate, dioxin, dimethylsulphoxide.
Postulated mode of action: Medroxy progesterone acetate (Fig. 4) is a progestin that is derived from the naturally occurring female hormone, progesterone. It is the derivative of 17α-hydroxyprogesterone (Pregnanes). It is used to treat abnormal uterine bleeding, promote menstrual cycles and to treat symptoms of the menopause. Medroxyprogesterone is use to promote menstruation when women do not begin naturally to menstruate at puberty (called primary amenorrhea) or if they stop menstruating before menopause (called secondary amenorrhea). It is used in the treatment of endometrial, prostate and renal cancer. It is also used for treating abnormal bleeding from the uterus in many situations. It is also used in combinations with estrogens for treating symptoms of menopause in order to prevent unchecked growth of the endometrium that may lead to endometrial cancer. It is secreted in breast milk, but the effect on infant has not been determined (Gilstrap and Little, 1992; Briggs et al., 2005). After oral intake medroxyprogesterone does not undergo any first pass effect. The bioavailability is 100%. It has no binding affinity to Sex-Hormone Binding Globulin (SHGB) and Corticosteroid Binding Globulin (CBG) and in serum medroxy progesterone acetate is bound to albumin for 88%. It is extensively metabolized in the liver (Schindler et al., 2003).
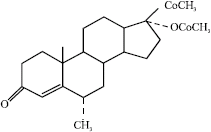 | |
| Fig. 4: | Medroxy progesterone acetate |
Cancer studies: Medroxyprogesterone acetate causes reversible changes in the endometrium, from proliferative to secretary or suppressed. Medroxyprogesterone acetate induced adeno carcinomas of the mammary gland in female mice and malignant mammary tumors in dogs. In female dogs a dose-related increase in the incidence of large mammary nodules was also found after intra-muscular administration (Lanari et al., 2001).
Genotoxic studies: Medroxyprogesterone acetate was reported to be negative in Ames test (Zeiger et al., 1992; Lang and Reimann, 1993) and V79 cells in vitro (Herzog and Leuschner, 1995). It was reported positive for unscheduled DNA synthesis in rat hepatocytes in vivo (Martelli et al., 2003), but negative in cultured human hepatocytes (Martelli et al., 2003). It does not induced micronucleus in mouse bone marrow cells (Morita et al., 1997) and chromosomal aberrations in rat bone marrow cells (Herzog and Leuschner, 1995). It did not form DNA adducts in human lymphocytes (Werner et al., 1997) and was reported to form DNA adducts in human liver slices in vitro (Feser et al., 1998). It induced chromosomal aberrations and sister chromatid exchanges in cultured human peripheral blood lymphocytes only in the presence of metabolic activation supplemented with NADP (Siddique et al., 2006a).
Chlormadinone acetate (CAS No. 302-22-7)
(6-chloro-17-hydroxypregna-4,6-diene-3, 20-dione acetate)
Colour: White or Creamy white
Odour: Odourless
Solubility: Chlormadinone acetate soluble in water, sparingly soluble in alcohol, soluble in acetone, ether and dimethysulphoxide.
Postulated mode of action: Chlormadinone acetate (Fig. 5) is a synthetic progesterone analogue. It is a derivative of 17α-hydroxyprogesterone (pregnanes), having chlorine atom at carbon-6 (Schindler et al., 2003). Chlormadinone acetate, with its anti-androgenic action (direct inhibitory effect on prostrate), exerts an inhibitory effect on hypertrophic prostrate, atrophic effects on prostrate. PROSTAL®-l Tablets contain 50 mg of chlormadinone acetate. When one tablet of PROSTAL-L Tablets was orally administrated to healthy male adults at the fasting state, the plasma concentration reached the maximum level at 5.1 h (Tmax) after the administration with a half life of 10.2 h (T½) showing the sustained release pattern of plasma concentration compared with ordinary chlormadinone acetate tablets. After oral intake chlormadinone acetate is rapidly absorbed and undergoes nearly no first pass metabolism. Therefore, the bioavailability is nearly 100%. It has no binding affinity to Estrogen Receptor (ER), Mineralocorticoid Receptor (MR) sex-hormone binding receptor and Corticosteroid-Binding Globulin (CBG). Chlormadinone acetate elimination occurs slowly. After 7 days only 34% of the dose is extracted. The reduction of the 3-keto-group results in the inactivation. 3-hydroxy-chlormadinone acetate is the important metabolite, which shows 70% of the anti-androgenic activity. Hydroxylation occurs at positions C2α, C3β and C15β. The majority of the metabolites are excreted renally, predominantly as glucoronides (Schindler et al., 2003). In cattle Chlormadinone is used for oestrus synchronizations at daily oral doses of 12 mg per animal for upto 20 days. The minimal effective level in human was 50 μg day-1 (CVMP, 200). Chlormadinone acetate is also used in sheep and goats for the same indication at daily oral doses of 2.5 mg per animal and in horses at daily oral doses of 12 mg per animal for upto 20 days. Its oral LD50 is 6400 mg kg-1 b.wt. but intraperitoneal LD50 in mice is 90 mg kg-1 b.wt. (Siddique and Afzal, 2004a).
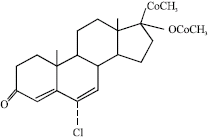 | |
| Fig. 5: | Chlormadinone acetate |
Cancer studies: Chlormadinone produced mammary tumors in dogs and also increase the incidence of mammary gland hyperplasia and mammary nodules (El Etreby and Graf, 1979). There is no data available on the genetic and related effects of chlormadinone acetate alone in humans.
Genotoxic studies: It was found negative in bacterial test system, unscheduled DNA synthesis (UDS) in rat hepatocytes in vitro (Topinka et al., 1995) and chromosomal aberrations in human peripheral blood lymphocytes in vitro (Stenchever et al., 1969), but in other study was reported to induced chromosomal aberrations and sister chromatid exchanges in cultured human lymphocytes (Siddique and Afzal, 2004b, 2005a; Siddique et al., 2008a). It was reported to form DNA adducts in rat liver in vitro (Topinka et al., 1995; Feser et al., 1996; Brambilla and Martelli, 2002) and human hepatocytes in vitro (Topinka et al., 1995; Werner et al., 1997) and micronucleus in rat liver cells in vivo (Martelli et al., 1996).
Ethynodiol diacetate CAS No. (297-76-7)
Colour: White or almost white
Odour: Odourless or almost odourless
Solubility: It is practically insoluble in water, soluble in alcohol, chloroform, ether and dimethyl sulphoxide. It is sparingly soluble in fixed oils.
Postulated mode of action: Ethynodiol diacetate (Fig. 6) is a derivative of 19-nortestosterone (estranes). It is used in the treatment of hypermenorrhea (menorrhagia), pain associated with endometriosis, dysmenorrhoea and dysfunctional uterine bleeding. It is also used as a female contraceptive. Ethynodioldiacetate tricks the body processes into thinking that ovulation has already occurred, by maintaining high levels of the synthetic progesterone. This prevents the release of eggs from ovaries. Ethynodiol diacetate and ethinylestradiol combination is used in oral contraceptives i.e., Demulen® and Zovia®. This combination prevents pregnancy by preventing ovulation (egg release). It causes a variety of hormonal changes. To prevent pregnancy after intercourse, the medicine either prevents or delays ovulation (egg release). Ethynodiol diacetate binds to the progesterone and estrogen receptors target cells include the female reproductive tract, mammary gland, the hypothalamus and the pituitary. Once bound the receptor, it shows the reduction in the frequency of release of Gonadotropin Releasing Hormone (GnRH) from the hypothalamus and blunt preovlatory LH (Lutenizing hormone) Surge. It is metabolized in liver or gut wall, to norethisterone and to sulphate and glucoronide conjugates. Its half life is about 25 h.
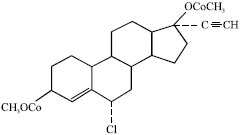 | |
| Fig. 6: | Ethynodiol diacetate |
Cancer studies: Ethynodioldiacetate increased the incidence of benign liver tumours in male mice and of mammary tumours in castrated male mice. There is no data available on the genetic and related effects of ethynodiol diacetate alone in humans. Oral administration of ethynodiol diacetate; in combination with mestranol to mice, increased the incidence of pituitary tumours. Ethynodiol diacetate plus ethinylestradiol increased the incidence of pituitary tumours and of malignant tumours of connective tissues of the uterus in mice. In rats it produced mammary tumours.
Genotoxic studies: Ethynodiol diacetate was reported to induced chromosomal aberrations and sister chromatid exchanges on cultured human lymphocyte in the presence of metabolic activation supplemented with NADP (Siddique and Afzal, 2004c; Siddique et al., 2007a).
Megestrol acetate (CAS: 595-33-5)
(17α-(Acetyloxy)-6-methyl-pregna-4,6-diene-3,20 dione)
Colour: White or creamy white
Odour: Odourless or almost odourless
Solubility: Megestrol acetate is practically in soluble in water, sparingly soluble in alcohol, very soluble in chloroform, slightly soluble in ether and in fixed oils.
Postulated mode of action: Megestrol acetate (Fig. 7) is a derivative of 17α-hydroxy progesterone (Pregnanes). It is used in oral contraceptives, breast and in the treatment of endometrial cancer. After oral intake the bioavailability of megestrol acetate is 100%. It is not bound to Sex Hormone Binding Protein (SHBG) or Corticosteroid Binding Globulin (CBG). It is bound to serum albumin. The most important metabolic pathways are hydroxylation reactions. It is metabolized by liver to glucoronide conjugates. It is excreted as conjugates metabolites via the urine and faeces (Schindler et al., 2003).
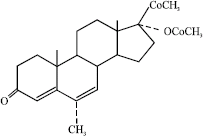 | |
| Fig. 7: | Megestrol acetate |
Cancer studies: Megestrol acetate plus ethinylestradiol has also been tested for carcinogenicity by oral administration in mice and rats. In mice, an increased incidence of malignant mammary tumours was observed in both sexes, but no increase in incidence of tumours was seen in rats. There is no data available on the genetic and related effects of megesterol acetae alone in humans (Joosten et al., 2004).
Genotoxic studies: It has been reported negative in unscheduled DNA synthesis test using rat hepatocytes; however, the presence of DNA adducts has been shown in rat liver in vivo and cultured human hepatocytes (Topinka et al., 1995; Feser et al., 1996; Werner et al., 1997). It has also been shown to induce micronucleus in rat liver in vivo, but has failed to cause chromosomal aberrations in human peripheral blood lymphocytes in vitro (Stenchever et al., 1969; Martelli et al., 1996). It induced chromosomal aberrations and sister chromatid exchanges in mice bone marrow cells (Siddique et al., 2005a).
Cyproterone acetate (CAS No: 427-51-0)
Colour: White or creamy white
Odour: Odourless or almost odourless
Solubility: Cyproterone acetate is practically insoluble in water, sparingly soluble in alcohol, very soluble in chloroform, acetone and dimethysulphoxide acetone, slightly soluble in ether and in fixed oils.
Postulated mode of action: Cyproterone acetate (Fig. 8) is a derivative of 17α-Hydroxyprogesterone (Pregnanes). In addition to the 6, 7 double bond, the 1,2 α-methyl group is present. Cyproterone acetate is a potent steroidal antiandrogen with progestational activity. It is used alone or in combination with ethinylestradiol or estradiol valerate in the treatment of women suffering from disorders associated with androgenization, e.g., acne or hisuitism. Cyproterone acetate competes with dihydrotestosterone for the androgen receptor and inhibits translocation of the hormone receptor complex in to the cell nucleus (Sciarra et al., 1990). The bioavailablity is nearly 100%. Cyproterone acetate has no binding affinity to sex hormone binding globulin and corticosteroid binding globulin in the serum but 93% of compound is bound to serum albumin. It is stored in fat tissue and excreted slowly. The important metabolic steps are hydroxylation reaction and de-acetylation. The metabolite 15β hydroxycyproterone acetate shows only 10% of the progestogenic potency of cyproterone itself. The bio-activation of the cyproterone acetate involves the reduction of the keto group at carbon-3, which is followed by sulfonation of the hydroxy steroid. The resulting sulfoconjugate is supposed to be very unstable and can decompose to a reactive DNA binding carbonium ion (Schindler et al., 2003).
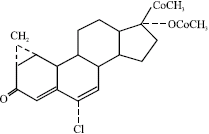 | |
| Fig. 8: | Cyproterone acetate |
Cancer studies: Cyproterone acetate is a tumour initiating agent in the liver of female rats (Deml et al., 1993).
Genotoxic studies: Cyproterone acetate was found negative in V79 cells in vitro (Lang and Reimann, 1993; Kasper et al., 1995) in Ames test (Lang and Reimann, 1993) and for micronucleus in mouse bone marrow cells in vivo (Reimann et al., 1996; Martelli et al., 1996) but it was positive for micronucleus in rat liver in vivo, for chromosomal aberrations in V79 cells (Kasper et al., 1995) and the human peripheral blood lymphocytes in vitro (Reimann et al., 1996). It induces mutation in Big Blue rat (Krebs et al., 1998). It was also found positive for unscheduled DNA synthesis (UDS) test in rat hepatocytes (Neumann et al., 1992; Kasper et al., 1995; Topinka et al., 1995; Martelli et al., 1995), human hepatocytes in vitro and in rat liver in vivo (Kasper and Muller, 1996) In female rats, DNA adducts have been observed at low doses of cyproterone acetate, which are in the range of therapeutic doses used in women (Werner et al., 1997). It was found to induce chromosomal aberrations and sister chromatid exchanges in human lymphocytes in vitro (Siddique and Afzal, 2005b, 2004d; Siddique et al., 2006b, 2007b, 2008b).
Lynestrenol (CAS No 52-76-6) (17α)-19-Norpregn-4-en-20yn-17ol)
Colour: White
Odour: Odourless
Solubility: Lynestrenol is practically insoluble in water, sparingly soluble in alcohol, very soluble inacetone, chloroform, dimethysulphoxide.
Postulated mode of action: Lynestrenol (Fig. 9) is used as single cavity drug or in combination with estrogen, such as ethinylestradiol or mestranol in oral contraceptives (Siddique and Afzal, 2004e). It is the derivative of 19-nortestosterone. Lynestrenol is converted in vivo to norethisterone. It is metabolized by 3β-hydroxylation and dehydrogenation (Schindler et al., 2003).
Cancer studies: The high doses of lynestrenol were associated with the increases incidence of mammary nodules and carcinomas (Misdorp, 1991). The LD50 value for lynestrenol for mice is 110 mg kg-1 body weight (Siddique and Afzal, 2005c).
Genotoxic studies: Lynestrenol induced chromosomal aberrations and sister chromatid ecology at the dosages of 13.75 and 27.50 mg kg-1 body weight in mice bone marrow cells (Siddique and Afzal, 2005c). It induced CA and SCE in cultured lymphocyte only in presence of metabolic activation (Siddique and Afzal, 2004e).
Norgestrel (CAS : 797-63-7)
(13β-Ethyl-17α-ethynyl-17β-hydroxygon-4en-3-one)
Colour: White or almost white
Odour: Odourless
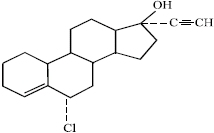 | |
| Fig. 9: | Lynestrenol |
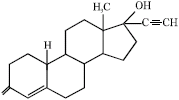 | |
| Fig. 10: | Norgestrel |
Solubility: Norgestrel is practically insoluble in water, sparingly soluble in alcohol, methylene chloride and freely soluble in chloroform and dimethyl sulphoxide.
Postulated mode of action: Norgestrel (Fig. 10) is the derivative of 19-nortestosterone (gonanes). It is a combination of both active and inactive enantiomers of which only the levorotary form is biologically active. It is used as a oral contraceptive either as single agent or in combination with an estrogen. Norgestrel was approved by the FDA in 1973. Norgestrel after oral administration is readily absorbed from the gastrointestinal tract and is widely distributed in body fluids. Protein binding is > 90% and is primarily to Sex Steroid Binding Globulin (SSBG) and albumin. Its half life is 10.26 h. Metabolism is believed to be hepatic with an elimination half life of about 20 h. Elimination is mostly via the urine with minimal amounts excreted in the bile and milk. Approximately 0.1% of the daily dose passes into breast milk (Schindler et al., 2003).
Cancer studies: It is reported to increase endometrial thickness during intrauterine progesterone therapy (Kresowik et al., 2008).
Genotoxic studies: It was reported to be genotoxic in cultured human peripheral blood lymphocyte with and without metabolic activation (Ahmad et al., 2001) and was negative for micronucleus in mouse bone marrow cells (Jordan, 2002). Norgestrel induced chromosomal aberration and sister chromatid exchanges in the presence of metabolic activation supplemented with NADP in culture human lymphocytes (Siddique et al., 2006c, 2007c).
Norethisterone (CAS No : 68-22-4)
(17α)-17-hydroxy-19-norpregn-4en-2oyn-3-one)
Colour: White or yellowish white
Odour: Odourless
Solubility: Norethisterone is insoluble in water, sparingly soluble in alcohol and soluble in chloroform and dimethylsulphoxide.
Postulated mode of action: Norethisterone (Fig. 11) is also named norethindrone and is often used as norethisterone acetate (NETA). Both compounds are rapidly absorbed from the gastro intestinal tract. The bio availability is about 64; 36% are bound to sex hormone binding globulin; 61% to serum albumin and 3% are free in circulation. Its half life is 5-12 h and is metabolised in the intestinal wall and liver. The principal metabolite is 5-α-dihydronorethisterone (Schindler et al., 2003).
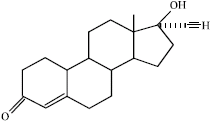 | |
| Fig. 11: | Northisterone |
Cancer studies: Norethisterone and its acetate were tested by oral administration in mice and rats and by subcutaneous implantation in mice. In mice, norethisterone and its acetate increased the incidence of benign liver cell tumours in males. It increased the incidence of pituitary tumours in females and produced granulosa cell tumours in the ovaries of females. It also increased the incidence of benign liver cell tumours and benign and malignant mammary tumours in male rats (Ahmad et al., 2001).
Genotoxic studies: Northisterone has been found to be negative in bacterial tests (Lang and Reimann, 1993; Dhillon and Dhillon, 1996). The widely used contraceptive steroid has been studied with respect to SCE and UDS induction and DNA adducts formation, in a range of in vitro and in vivo systems. Induction of UDS in human and rat hepatocytes was either positive or variable (Martelli et al., 2003). A metabolic gender difference was found : male but not female hepatocytes gave UDS response (Joosten et al., 2004), 32-P-post labelling was negative in rat liver in vivo (Feser et al., 1996) SCE induction in human lymphocytes was negative in one study (Ahmad et al., 2001), but positive in another study (Dhillon and Dhillon, 1996). Administration of norethisterone to mice caused SCE formation in bone marrow cells (Dhillon and Dhillon, 1996). Positive, negative and variable results have been obtained in tests for chromosomal aberrations or micronucleus induction with norethisterone (Dhillon and Dhillon, 1996; Stenchever et al., 1969; Martelli et al., 1998).
Norethynodrel (CAS No : 68-23-5)
(17 α)-17-hydroxyl-19-norpregn-5 (10) en-20yn-3-one)
Colour: White or almost white
Odour: Odourless
Solubility: Practically insoluble in water; soluble in alcohol, chloroform, dimethylsulphoxide.
Cancer studies: Norethynodrel (Fig. 12) was tested by oral administration in mice and rats and by subcutaneous implantation in mice. It increased the incidence of pituitary tumours in mice of each sex and that of mammary tumours in castrated mates of one strain. It also increased the incidence of benign and malignant liver cell, pituitary and mammary (benign and malignant) tumours in male rats (Jordan et al., 1993).
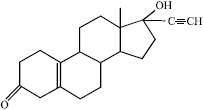 | |
| Fig. 12: | Norethynodrel |
Genotoxic studies: Norethynodrel was negative for ames test (Lang and Reimann, 1993). For unscheduled DNA synthesis it was positive in male rat hepatocytes in vitro, but negative in rat hepatocytes in vitro (Joosten et al., 2004). Norethynoderel did not induced aneuploidy in human cells in culture or unscheduled DNA synthesis in rat hepatocytes in vitro it inhibited inter cellular communications in Chinese hamster V79cells (Joosten et al., 2004). It induced chromosomal aberrations and sister chromatid exchanges in the presence of metabolic activation supplemented with NADP in cultured human lymphocytes (Siddique and Afzal, 2005d; Siddique et al., 2007d, 2006d) and chromosomal aberrations and sister chromatid exchanges at 15.62 and 31.25 mg g-1 body weight in mice bone marrow cells with a LD 50 dose of 125 mg kg-1 (Siddique and Afzal, 2003).
Dimethisterone (CAS No : 79-64-1)
(6α,17β)-17-hydroxyl-6-methyl-17-(1-propynyl)-adrst-4en-3-one)
Colour: White
Odour: Odourless
Solubility: It is insoluble in water, freely soluble in dehydrated alcohol, very soluble in chloroform, acetone, dimethyl sulphoxide.
Cancer studies: Dimethisterone (Fig. 13) was tested for its carcinogenicity in various animal models. There is no increase in tumour incidence (Drill, 1980).
Genotoxic studies: It did not induced chromosomal aberration in cultured human peripheral blood lymphocytes (Stenchever et al., 1969).
Progesterone (CAS No : 57-83-0)
(Preg-4-ene-3, 20 dione)
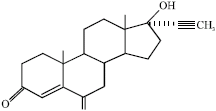 | |
| Fig. 13: | Dimethisterone |
Colour: White or slightly yellowish white
Odour: Odourless
Solubility: It is practically insoluble in water, freely soluble in dehydrated alcohol, very soluble in chloroform, sparingly soluble in acetone, in ether, in fixed oils and dimethysulphoxide.
Postulated mode of action: Progesterone (Fig. 14) is used in oral contraceptives and in cure of premenstrual syndrome. It is 95-98% bound to plasma proteins and is metabolized as glucoronide conjugate by the liver (Schindler et al., 2003).
Cancer studies: Progesterone was tested by subcutaneous and intra by intramuscular injection in mice, rabbits and dogs and by subcutaneous implantation in mice. It increased the incidences of ovarian, uterine and mammary tumours in mice. Neonatal treatment with progesterone enhanced the occurrence of pre-cancerous and cancerous lesions of the genital tract and increased mammary tumorigenesis in female mice (Lamb et al., 2007).
Genotoxic studies: Progesterone did not induce dominant lethal mutations in mice or chromosomal aberrations in rats treated in vivo. It did not induced chromosomal aberration or sister chromatid exchange in cultured human cells, nor chromosomal aberrations or DNA strand breaks in rodent cells. Studies on transformation of rodent cells in vivo were inconclusive; a clearly positive result was obtained for rat embryo cells, are weakly positive result for mouse cells and a negative result for Syrian Hamster embryo cells. Progesterone was not mutagenic to bacteria (Joosten et al., 2004) The test results of the L5178-YTK mouse lymphoma assay were inconclusive (Myhr and Caspary, 1988), All data a UDS induction in male rat or human hepatocytes (Oshiro et al., 1986; Martelli et al., 2003) or 32P-post labelling in the rat liver in vivo or human hepatocytes (Feser et al., 1996; Werner et al., 1997), indicate that progesterone does not cause DNA lesions variable responses with inter-animal differences in UDS were observed in male and female rat hepatocytes (Martelli et al., 2003). Progesterone was not clastogenic in cultured human lymphocytes (Stenchever et al., 1969).
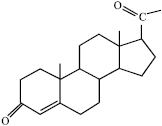 | |
| Fig. 14: | Progesterone |
Increased frequencies of micronuclei were found in hepatocytes of rats treated with a single oral dose (100 mg kg-1) of progesterone (Martelli et al., 1998). It does not induce an euploidy in DON cells in vitro (Wheeler et al., 1986).
MECHANISM FOR THE GENTOTOXICITY
The carcinogenic effects of hormone replacement therapy used to relieve symptoms of menopause were evaluated by the International Agency for Research on Cancer (Joosten et al., 2004). Most of the studies reviewed did not differentiate between the effects of estrogen only and estrogen-progestin combination therapies. An increased risk of endometrial cancer was associated with increasing duration of therapy (Kresowik et al., 2008). Numerous case control and cohort studies have addressed the risk of various cancers associated with the use of oral contraceptives (Heinemann et al., 1997). Most of the studies involved estrogen-progestins combinations. Studies in rats, mice, hamsters and guinea pigs have been conducted with estrogens alone or in combination with known carcinogens. Estrogen had a carcinogenic effect in all species and by all routes of administration. Most studies showed induction of benign and malignant neoplasias as well as preneoplastic lesions, in a variety of target organs, including the breast and female reproductive tract (Joosten et al., 2004).
Steroidal estrogens can damage chromosomes and DNA in mammals (Banerjee et al., 1994; Siddique and Afzal, 2004f; Siddique et al., 2005b). The most frequently reported effects include DNA adduct formation, cytogenetic alterations (e.g chromosome and chromatid breaks, micronuclei, SCEs), aneuploidy and cell transformation (Djelic and Djelic, 2002). The genotoxic effects of estrogens/synthetic progestins/androgens have been demonstrated in various in vitro assays, using cultured animal cells or cell free systems. Fewer effects have been reported in whole animal studies or in studies with human cells and no human in vivo studies were identified (Siddique et al., 2007e; Beg et al., 2007; Joosten et al., 2004; Djelic et al., 2005).
Many different formulations of synthetic and naturally produced estrogens are prescribed for use as oral contraceptives or in postmenopausal hormone replacement therapy (Biri et al., 2002). Exogenous estrogens are well absorbed from the gastro intestinal tract and the skin of human animals. Estrogens are metabolized in the gastro intestinal and other tissues. In both human and animals, estrogens undergo similar phase I and Phase II reactions. Aromatic hydroxylation reactions catalyzed by cytochrome P-450 enzymes are the primary phase 1 pathways. (Chen et al., 1998). Sulfation, methylation and glutathione conjugation are the major phase 2 pathways. The major phase 1 metabolic pathway for endogenous estrogens is aromatic hydroxylation to catechol intermediates. Concerning the genotoxic effects of estrogens; 16α-Hydroxyestrone, 4-hdroxyestradiol and 4-hydroxyestrone have direct genotoxic effects and carcinogenicity. Liehr et al. (1986) described mechanistic similarities between human breast cancer and estrogen induced kidney cancer in hamsters and identified metabolism to the 4-hydroxylated catechols as the primary pathway leading to tumour development. The 4-hydroxylated catechols may undergo subsequent redox cycling between semiquinone and quinone forms. The quinones may undergo nonenzymatic isomerization to quinone metabolites. The quinone and quinone metabolites intermediates are highly reactive and may form covalent DNA adducts, thus these metabolites are candidates for ultimate estrogen carcinogens. Redox cycling between various forms of quinones generates superoxide radicals that are capable of direct and indirect damage to DNA. In breast cancer cells, 4-hydroxylation predominates over 2-hydroxyltion (Zhu and Conney, 1998).
Excessive production of reactive oxygen species has been reported in breast cancer tissue and free radical toxicity (DNA single strand breaks, lipid peroxidation and chromosomal abnormality) has been reported in hamsters treated with estradiol. Reactive oxygen species, including superoxide, hydrogen peroxide and hydroxyl radicals, may be produced through redox cycling between the O-quinones and their semi quinone radicals (Roy et al., 1991). These reactive oxygen species can cause oxidative cleavage of the phosphate-sugar back bone and oxidation of the purine and pyrimidine residues of DNA. The incubation of 4-hydroxylated catechols with microsomes, NADPH and DNA resulted in 8-hydroxylation of guanine bases. 8-hydroxy-deoxyguanosine is a biomarker for oxidative damage and is considered an important factor in carcinogensis (Han and Liehr, 1994; Bolton et al., 1998; Bolton, 2002). The possible mechanism and cause of the genotoxicity at different dosages of synthetic progestins such as cyproterone acetate, Chlormadinone acetate, medroxy progesterone acetate, have been studied by using different doses of superoxide dismutase and catalase in the presence of metabolic activation with and without NADP. Superoxide dismutases (SODs) are family of metal enzymes that convert O.-2 to H2O2 according to the following reaction (Culotta, 2000).
Catalase is a heme containing protein that brings about the decomposition of H2O2 into water and oxygen. Catalase is found to reduce sister chromatid exchanges levels. The mechanism of reaction is (Reid, 2003):
The treatment of SOD with ethinylestradiol in the presence of metabolic activation with NADP has been reported to increase the genotoxic damage (Siddique et al., 2005b). Since ethinyl estradiol is genotoxic only in the presence of metabolic activation with NADP, the first step would involve the aromatic hydroxylation catalysed by cytochrome P450, as occurs in the case of estrone and the 17β-estradiol forming catechol metabolites (Yager and Liehr, 1996; Bolton et al., 1998). Cytochrome P450, in liver S9 fractions plays an important role in activating promutagens to proximated/or ultimate mutagens. Rat and human liver P450 is involved in the activation of some chemical carcinogens having different isoforms (Maron and Ames, 1983; Guengerich and Shimada, 1991). In the tissues of liver, 2-hydroxylation predominates over 4-hydroxylation by approximately 9:1, however, in extra hepatic tissues the ratio drops to 1:1 (Zhu and Conney, 1998). Only 4-hydroxyestrone-3, 4-dihydroxy 1,3,5 (10)-oestraien-17one (4-OHE) was found to be carcinogenic in the male Syrian golden hamster kidney tumour model, whereas 2-OHE was found to be without any activity (Yager and Liehr, 1996; Guengerich and Shimada, 1991). Once formed, the endogenous catechol estrogens can be oxidized by virtually any oxidative enzymes in the absence or presence of metal ion and can give rise to o-quinones (Yager and Liehr, 1996; Bolton et al., 1998). The study on ethinylestradiol confirms the presence of Reactive Oxygen Species (ROS) and the source of reactive oxygen species has been suggested to be the result of redox cycling between o-quinones and their semiquinone radicals, generating superoxide, hydrogen peroxide and ultimately reactive hydroxyl radicals, that cause oxidative cleavage of the phosphate sugar backbone as well as oxidation of the purine/pyrimidine residues of DNA (Han and Liehr, 1994).
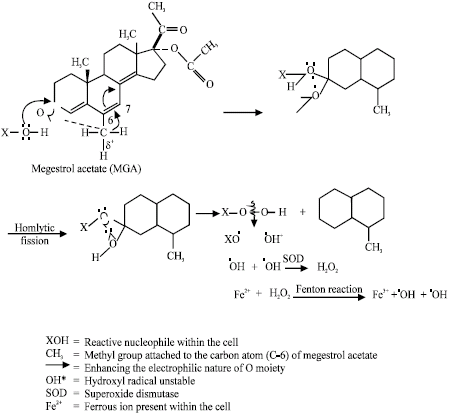 | |
| Fig. 15: | Possible mechanism of generating free radicals by megestrol acetate (MGA) (Siddique et al., 2005a; Hum Exp Toxicol) |
The study on cyproterone acetate, chlormadinone acetate and megestrol acetate reveal that the compounds increases chromosomal aberrations and frequencies of sister chromatid exchange, significantly in cultured human lymphocytes (Siddique and Afzal, 2004b, 2005a, b). The treatment with superoxide dismutase, increased the frequencies of chromosomal aberrations and sister chromatid exchanges generated by both cyproterone acetate and chlormadinone acetate. The possible mechanism of free radical generation for cyproterone acetate, megestrol acetate and chlormadinone acetate is shown in Fig. 15-17.
XOOH can also give rise XOO• (alkoxyl) and •OOH (Peroxyl) radicals. The presence of alkoxyl (XOO•) radical appears to be a very remote possibility in view of the highly polar nature of the living system, because X would have to be essentially an alkyl group (Islam et al., 1991). Hydroxyl radical (OH) is converted into hydrogen peroxide by superoxide dismutase (Culotta, 2000), as a result, the increase in chromosomal aberrations and sister chromatid exchanges was observed when the cyproterone acetate and chlormadinone acetate were treated with superoxide dismutase. The catalase treatment with cyproterone acetate and chlormadinone acetate have been reported reduce the genotoxic damage (Siddique and Afzal, 2004b, 2005a, b).
Megestrol acetate has been reported to be genotoxic in mice bone marrow cells (Siddique et al., 2005a). The treatment of megestrol acetate with ascorbic acid reduced the genotoxic damage of megestrol acetate. Ascorbic acid posses a substantial nucleophilic character and it has been suggested that ascorbate might protect against electrophilic attack a cellular DNA by intercepting reactive agents (Edgar, 1974). Reactive oxygen species generated by MGA via nucleophilic reaction have been suggested responsible for the genotoxic damage (Siddique et al., 2005a). Norgestrel shows genotoxic effects only in the presence of metabolic activation in the presence of NADP (Siddique et al., 2006c). The metabolic activation of norgestrel and possible conversion of it to reactive species may be responsible for its genotoxicity. Metabolic activation of estrogens such as estradiol-17β and ethinyl estradiol results in the production of reactive oxygen species via redox cycling between quinones and semi-quinone radicals (Fig. 18) (Siddique et al., 2005b).
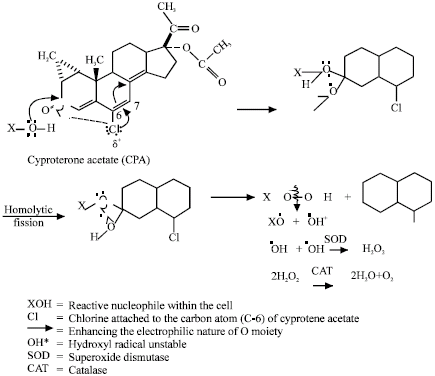 | |
| Fig. 16: | Possible mechanism of generating free radicals by cyproterone acetate (Siddique and Afzal, 2005b; Toxicol in vitro) |
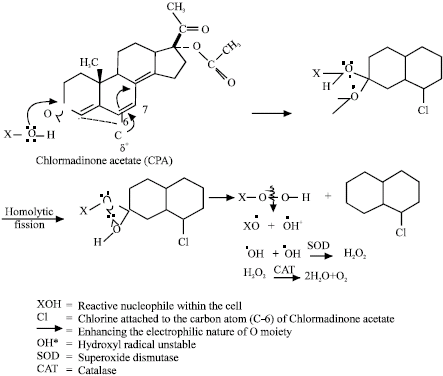 | |
| Fig. 17: | Possible mechanism of generating reactive oxygen species by chlormadinone acetate (Siddique and Afzal, 2004b) Indian J Exp Biol |
Medroxyprogesterone acetate (Fig. 19) showed genotoxic effects in the presence of metabolic activation supplemented with NADP (Siddique et al., 2006a).The genotoxic potential of various synthetic progesting has been tested in different experimental models using different genotoxic end points (Joosten et al., 2004). Very little attention has been paid to the structural relationship and the potentiality to induce the genotoxic damage by synthetic progestins. Structural relationship and ability to induce genotoxic damage has been worked out, using DNA repair assay and micronucleus formation in the liver of female rats (Brambilla and Martelli, 1997). For DNA repair following trend was found, viz. cyproterone acetate > chlormadinone acetate > megestrol acetate. It was reported that cyproterone acetate with (a) a1, 2α-methylene group, (b) a keto group at carbon-3, (c) two double bonds, C4 = C5 and C6 = C7 and (d) Cl at carbon-6, is the most genotoxic molecule. The lower genotoxic potencies of chlormadinone acetate and megestrol acetate might well be due to the absence of 1, 2α-methylene group and for megestrol acetate, perhaps due to the presence of methyl group (-CH3) at carbon-6 instead of Cl (Brambilla and Martelli, 1997). However, when the adduct formation in cyproterone acetate, medroxyprogesterone acetate, megestrol acetate and chlormadinone acetate are considered, the cyproterone acetate, megestrol acetate and chlormadinone acetate are found to form adducts in human liver slices in vitro. Medroxyprogesterone acetate forms these at low level (Feser et al., 1998).
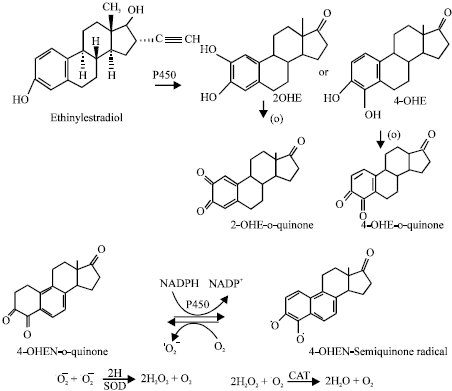 | |
| Fig. 18: | Possible metabolism ethinylestradiol to catechols and quinones and generation of reactive oxygen species by redox cycling of 4-hydroxy equilenin (Siddique et al., 2005b, Chem Biol Interact) |
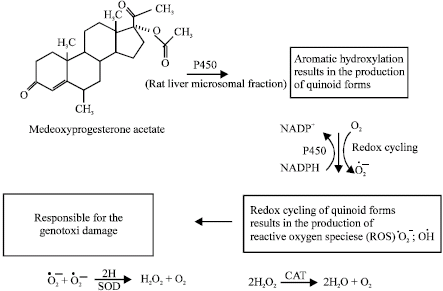 | |
| Fig. 19: | Possible metabolism and generation of reactive oxygen species by medroxyprogestrone acetate (Siddique et al., 2006a Life Sci) |
Studies performed on megestrol acetate (Siddique et al., 2005a), cyproterone acetate (Siddique and Afzal, 2005b), chlormadinone acetate (Siddique and Afzal, 2004b), ethynodiol diacetate (Siddique and Afzal, 2004c), norethynodrel (Siddique and Afzal, 2005d), norgestrel (Siddique et al., 2006c) and lynestrenol (Siddique and Afzal, 2004e), show that the progestins, in which double bond between carbon-6 and carbon-7 is present, like megestrol acetate, cyproterone acetate and chlormadinone acetate, can undergo nucleophilic reaction and generate free radicals in the system and therefore, show the genotoxic effects and the progestins, in which double bond between carbon-6 and carbon-7 is absent, may require additional metabolic activation to show the genotoxic effects in the test system. Looking at the chemical structure of estradiol and ethinylestradiol, the double bond is found to be absent between carbon-6 and carbon-7. It is well known that estradiol undergoes aromatic hydroxylation in liver by cytochrome P450s and results in the formation of catechol metabolites (Maclusky et al., 1981; Fishman, 1983; Liehr et al., 1986; Li and Li, 1987; Bolton, 2002). The redox cycling between O-quinones and their semi-quinone radicals results in the generation of superoxide, hydrogen peroxide and ultimately reactive hydroxyl radicals which cause oxidative cleavage of the phosphate-sugar backbone as well as oxidation of the purine/pyrimidine residues of DNA (Han and Liehr, 1994). Ethinylestradiol generates reactive oxygen species in the presence of metabolic activation with NADP (Siddique et al., 2005b). The study performed on norgestrel, ethynodioldiacetate, lynestrenol and norethynodrel shows that they exhibit genotoxic effects only in the presence of metabolic activation with NADP. The similarity in the structure of the above synthetic progestins and the estrogens lies mainly in the absence of double bond between carbon-6 and carbon-7 (Fig. 20).
Moreover, if the adduct formation on the basis of presence and absence of double bond between carbon-6 and carbon-7 is considered, a definite correlation is established. A study, carried out by Feser et al. (1998) on some selected sex-steroids, shows that chlormadinone acetate (C6 = C7) and megestrol acetate (C6 = C7) from DNA adducts, while norgestrel (C6-C7), estradiol (C6-C7) and ethinylestradiol (C6-C7) and ethinylestradiol (C6-C7) do not form DNA adducts in human liver slices. it is thus concluded that the double bond between carbon-6 and carbon-7 plays an important role in determining the mode of the genotoxicity of synthetic progestins. On the basis of above information the model for the genotoxicity of steroids (synthetic progestins/estrogens) is suggested as shown in the Fig. 21. Estrogens have been reported to induce genotoxicity in various in vitro and in vivo models. Synthetic progestins are at second position. Estrogens caused genotoxicity by metabolic activation i.e., forming catechol estrogens. The balance of toxification and detoxification determines the responsiviness by an individual and it is species and tissue dependent. The natural estrogen can cause DNA damage in the following ways (Joosten et al., 2004).
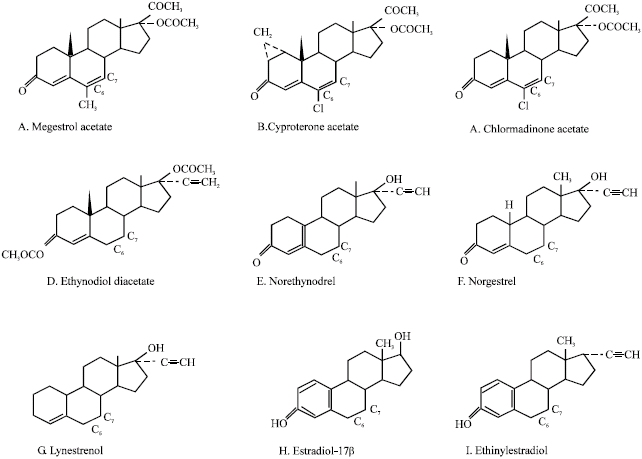 | |
| Fig. 20: | Structure of synthetic progestins. Presence of double bond between carbon-6 and carbon-7 (A to C. Absence of double bond between carbon-6 and carbon-7 (D-I). (Siddique et al., 2007c; Biomed Res) |
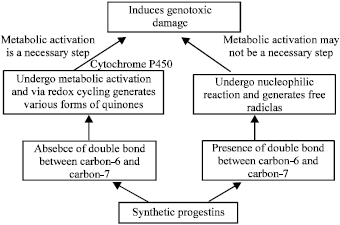 | |
| Fig. 21: | A model for the genotoxicity of synthetic progestins |
| • | Redox cycling of catechol estrogen gives rise to reactive oxygen species (O2-) that can cause single strand break |
| • | Hydroxyl radical can oxidized DNA base (Guanine) |
| • | Lipid peroxidation may also take place and results in the formation of malonaldehyde DNA adducts |
| • | Estrogen-DNA adducts may be formed after metabolic activation |
For the synthetic progestin present study shows that the presence of double bond between carbon-6 and carbon-7 might be important for the genotoxicity. The presence of double bond is and the adduct formation is also correlated with the study performed on few selected synthetic progestins by Fesar et al. (1998). The contradictory results for the studied synthetic progestins may be due to the low robustness of the various test models (Clastogenicity). In vitro conditions may generally favour phase-I activation reactions while not allowing phase-II de-activation reactions. The metabolic activation is a necessary step for the genotoxicity of natural estrogens as well as for the synthetic progestins. Some synthetic progestins are also genotoxic in the absence of metabolic activation (Joosten et al., 2004). The dose is an important factor in determining the genotoxicity testing. Dose response relationship must be studied to reach any conclusion. Generally high doses of estrogens and synthetic progestins have been studied in various in vitro experimental models i.e., in μM and μg. The therapeutic plasma concentrations are often in the nanogram or pictogram mL-1 range. The high doses studied are significant because this dose range may reach in some clinical abnormal conditions or due to lack of metabolizing enzymes or other factors (Martelli et al., 2003). As it is evident from the epidemiological studies that prolonged users of steroids, are at risk of having various types of cancers. The dose factor and the clastogeny as its own importance. An increase in the frequency of chromosomal aberrations in peripheral blood lymhocytes is associated with an increase in overall risk of cancer (Hagmar et al., 1994, 1998).
CONCLUSION
The steroids are genotoxic only at high doses. The therapeutic doses are safe. The care should be taken with regard to their concentration as they may be carcinogenic in the long term use in humans.
ACKNOWLEDGMENTS
Thanks are due to the Chairman, Department of Zoology, Aligarh Muslim University, Aligarh for providing laboratory facilities. The research grant (No:09/112(0355)/2003-EMR-1) from the Council of Scientific and Industrial Research, New Delhi and Department of Science and Technology (SR/FT/LS-003/2007), New Delhi are also acknowledged.
REFERENCES
- Ahmad, M.E., G.G.H.A. Shadab, M.A. Azfer and M. Afzal, 2001. Evaluation of genotoxic potential of synthetic progestins norethindrone and norgestrel in human lymphocytes in vitro. Mutat. Res., 494: 13-20.
PubMed - Banerjee, S.K., S. Banerjee, S.A. Li and J.J. Li, 1994. Induction of chromosome aberrations in Syrian Hamster renal cortical cells by various estrogens. Mutat. Res., 311: 191-197.
PubMed - Beg, T., Y.H. Siddique and M. Afzal, 2007. Chromosomal damage induced by androgenic anabolic steroids, stanozolol and trenbolone, in human lymphocytes. Adv. Environ. Biol., 1: 39-43.
Direct Link - Biri, A., E. Civelek, B. Karahalil and S. Sardas, 2002. Assessment of DNA damage in women using oral contraceptives. Mutat. Res., 521: 113-119.
PubMed - Bolton, J.L., 2002. Quinoids, quinoid radicals and phenoxyl radicals formed from estrogens and antiestrogens. Toxicology, 177: 55-65.
PubMed - Bolton, J.L., E. Pisha, F. Zhang and S. Qiu, 1998. Role of quinoids in estrogen carcinogenesis. Chem. Res. Toxicol., 11: 1113-1127.
PubMed - Brambilla, G. and A. Martelli, 1997. Preliminary data for a study on relationship between structure and genotoxicity of progestins. Mutat. Res., 379: S15-S15.
Direct Link - Brambilla, G. and M. Martelli, 2002. Are some progestins genotoxic liver carcinogens?. Mutat. Res., 512: 155-163.
PubMed - Briggs, G.G., R.K. Freeman and S.J. Yaffe, 2005. Drugs in Pregnancy and Lactation: In: A Reference Guide to Fetal and Neonatal Risk. 1st Edn., Lippincott Williams and Wilkins, Baltimore, USA., ISBN 0781756510, pp: 1858.
Direct Link - Carr, D.H., 1970. Chromosome studies in selected spontaneous abortions.1.Conception after oral contraceptives. Can. Med. Assoc. J., 103: 343-348.
PubMed - Chang, M.C. and D.M. Hunt, 1970. Effects of various progestins and estrogens on the gamete transport and fertilization in the rabbit. Fertil. Ster., 21: 683-686.
PubMed - Chen, Y., L. Shen, F. Zang, S.S. Lau, R.B. Van Breeman, D. Nickolic and J.L. Bolton, 1998. The equine estrogen metabolite 4-hydroequilenin causes DNA single-strand breaks and oxidation of DNA bases in vitro. Chem. Res. Toxicol., 11: 1105-1111.
PubMed - Culotta, V.C., 2000. Superoxide dismutase, oxidative stress and cell metabolism. Curr. Top. Cell. Reg., 36: 117-132.
PubMed - Deml, E., L.R. Schwarz and D. Oesterle, 1993. Initiation of enzyme altered foci by the synthetic steroid cyproterone acetate in rat liver foci bioassay. Carcinogenesis, 14: 1229-1231.
PubMed - Dhillon, V.S. and I.K. Dhillon, 1996. Genotoxicity evaluation of norethisterone acetate. Mutat. Res., 367: 1-10.
PubMed - Djelic, N. and D. Djelic, 2002. Enanced sister chromatid exchange rate in human lymphocytes exposed to 17-β estradiol in vitro. Arch. Med. Res., 33: 148-151.
PubMed - Djelic, N., B.S. Potparevic and D. Djelic, 2005. Mutagenic activity of estradiol evaluated by an in vitro micro nucleus assay. Acta Biol. Hung., 56: 403-406.
PubMed - Drill, V.A., 1980. Relationship of estrogens and oral contraceptives to endometrial cancer in animals and women. J. Reprod. Med., 24: 5-13.
PubMed - El Etreby, M.F. and K.J. Gräf, 1979. Effect of contraceptive steroids on mammary gland of beagle dog and its relevance to human carcinogenicity. Pharmacol. Therap., 5: 369-402.
PubMed - Elstein, M., S.E. Morris, G.V. Groom, D.A. Jenner, J.J. Scarisbrick, E.H. Cameron, 1976. Studies on low dose oral contraceptives: Cervical and plasma hormone changes in relation to circulating d-norgestrel and 17-α ethinyl estradiol concentrations. Fertil. Ster., 27: 892-899.
PubMed - Goh, K.O., 1967. Chromosomal breaks in women taking birth control pills. USAEC-ORAU Res. Report, 106: 97-104.
Direct Link - Guengerich, F.P. and T. Shimada, 1991. Oxidation of toxic and carcinogenic chemicals by human cytochrome P-450 enzymes. Chem. Res. Toxicol., 4: 391-407.
CrossRefPubMedDirect Link - Hagmar, L., A. Brogger, I.L. Hansteen, S. Heims and B. Hoggstedt et al., 1994. Cancer risk in humans predicted by increased level of chromosomal aberrations in human lymphocytes: Nordic study group on the health risk of chromosome damage. Cancer Res., 54: 2919-2922.
Direct Link - Hagmar, L., S. Bonassi, U. Stromberg, A. Brogger, J.E. Knudson, H. Norppa and C. Reuterwall, 1998. Chromosomal aberrations in human lymphocytes predicts human cancer: A report from the European study group on cytogenetic biomarkersnd health (ESCH). Cancer Res., 58: 4117-4121.
PubMed - Han, X. and J.G. Liehr, 1994. 8-Hydroxylation of guanine bases in kidney and liver DNA of Hamsters treated with estradiol: Role of free radicals in estrogen induced carcinogenesis. Can. Res., 54: 5515-5517.
PubMed - Heinemann, L.A.J., T. Do Minh, I. Guggenmoos-Holzmann, C. Thie and E. Garbe et al., 1997. Oral contraceptives and liver cancer. Results of the multicentre international liver tumor study (MILTS). Contraception, 56: 275-284.
PubMed - Heinemann, L.A.J., T. Do Minh, I. Guggenmoos-Holzmann, C. Thie and E. Garbe et al., 1997. Oral contraceptives and liver cancer. Results of the multicentre international liver tumor study (MILTS). Contraception, 56: 275-284.
PubMed - Islam, S., Shafiullah and M. Ahmad, 1991. Mutagenic activity of certain synthetic steroids: structural requirement for mutagenic activity in Salmonella and E. coli. Mutat. Res., 259: 177-187.
PubMed - Joosten, H.F.P., F.A.A. van Acker, D.J. van den Dobbelsteen, G.J.M.J. Horbach and E.I. Krantc, 2004. Genotoxicity of hormonal steroids. Toxicol. Lett., 151: 113-134.
CrossRef - Jordan, A., 2002. Toxicology of progestogens of implantable contraceptives for women. Contraception, 65: 3-8.
PubMed - Jordan, V.C., M.H. Jeng, W.H. Catherino and C.J. Parker, 1993. The estrogenic activity of synthetic progestins used in oral contraceptives. Cancer, 71: 1501-1505.
PubMed - Kasper, P. and I. Müeller, 1996. Time related induction of DNA repair synthesis in rat hepatocytes following in vivo treatment with cyproterone acetate. Carcinogenesis, 17: 2271-2274.
PubMed - Kasper, P., K. Tegethoff and L. Müeller, 1995. In vitro mutagenicity studies on cyproterone acetate using female rat hepatocytes for metabolic activation and as indicator cells. Carcinogenesis, 16: 2309-2314.
PubMed - Krebs, O., B. Schafer, T. Wolff, D. Oesterle, E. Deml, M. Sund and J. Favor, 1998. The DNA damaging drug cyproterone acetate causes a gene mutations and induces glutathione-S-transferase in the liver of female Big BlueTM transgenic F 344 rats. Carcinogenesis, 11: 241-245.
PubMed - Kresowik, J., G.L. Ryan, B.J. and Van Voorhis, 2008. Progression of atypical endometrial hyperplasia to adenocarcinoma despite intrauterine progesterone treatment with the levonorgestrel-releasing intrauterine system. Obstet. Gynecol., 111: 547-549.
PubMed - Lamb, R., H. Harrison and R.B. Clarke, 2007. Mammary development, carcinomas and progesterone: role of Wnt signaling. Ernst Schering Found Symp. Proc., 1: 1-23.
PubMed - Lanari, C., I. Lüthy, C.A. Lamb, V. Fabris and E. Pagano et al., 2001. Five novel hormone-responsive cell lines derived from murine mammary ductal carcinomas: In vivo and in vitro effects of estrogens and progestins. Cancer Res., 61: 293-302.
PubMed - Lang, R. and R. Reimann, 1993. Studies for a genotoxic potential of some endogenous and exogenous steroids. I. Communication: Examination for the induction of gene mutations using the Ames Salmonella/Microsomes Test and the HGPRT Test in V79 cells. Environ. Mol. Mutagen., 21: 272-304.
PubMed - Li, J.J. and S.A. Li, 1987. Estrogen carcinogenesis in Syrian hamster tissues: Role of metabolism. Fed. Proc., 46: 1858-1863.
PubMed - Liehr, J.G., W.F. Fang, D.A. Sirbasku and A. Ari-Ulubelen, 1986. Carcinogenecity of catechol estrogens in Syrian Hamster. J. Steroid Biochem., 24: 353-356.
PubMed - Maclusky, N.J., F. Naftolin, L.C. Krey and S. Franks, 1981. The catechol estrogens. J. Steroid Biochem., 15: 111-124.
PubMed - Maier, W.E. and J.R. Herman, 2001. Pharmacology and toxicology of ethinylestradiol and norethindrone acetate in experimental animals. Regulatory Toxicol. Pharmacol., 34: 53-61.
PubMed - Maron, D.M. and B.N. Ames, 1983. Revised methods for the Salmonella mutagenicity test. Mutat. Res./Environ. Mutagen. Relat. Subj., 113: 173-215.
CrossRefDirect Link - Martelli, A., E. Mereto, M. Ghia, P. Orsi and A. Allavena et al., 1998. Induction of micronuclei and of enzyme-altered foci in the liver of female rats exposed to progesterone and three synthetic progestins. Mutat. Res., 419: 33-41.
PubMed - Martelli, A., F. Mattioli, M. Angiola, R. Reimann and G. Brambilla, 2003. Species, sex and interindividual differences in DNA repair induced by nine sex steroids in primary cultures of rats and human hepatocytes. Mutat. Res., 536: 69-78.
Direct Link - Martelli, A., F. Mattioli, S. Fazio, U. Andrae and G. Brambilla, 1995. DNA repair synthesis and DNA fragmentation in primary cultures of human and rat hepatocytes exposed to cyproterone acetate. Carcinogenesis, 16: 1265-1269.
PubMed - Martelli, A., G.B. Campart, M. Ghia, A. Allavena, E. Mereto and G. Brambilla, 1996. Induction of micronuclei and initiation of enzyme altered foci in the liver of female rats treated with cyproterone acetate, chlormadinone acetate or megestrol acetate. Carcinogenesis, 17: 551-554.
PubMed - Misdorp, W., 1991. Progestogens and mammary tumors in dogs and cats. Acta Endocrinol., 125: 27-31.
PubMed - Morita, T., N. Asano, T. Awogi, Y.F. Sasaki and S. Sato et al., 1997. Evaluation of the rodent micronucleous assa in the screening of IARC carcinogens (groups 1, 2 and 2B). The summary report of the 6th collaborative study of the micronucleus group test mammalian mutagenicity study group. Mutat. Res., 389: 3-122.
PubMed - Myhr, B.C. and W.J. Caspary, 1988. Evaluation of the L 5178 Y mouse lympho cell mutagenesis assay: Iresults for sixty-three coded chemicals tested at Litton Biometric Inc. Environ. Mol. Mutagen., 12: 103-194.
PubMed - Murthy, P.B. and K. Prema, 1979. Sister chromatid exchanges in oral contraceptives users. Mutat. Res., 68: 149-152.
PubMed - Neumann, I., D. Thierau, U. Andrae, H. Greim and L.R. Schwarz, 1992. Cyproterone acetate induces DNA damage in cultured rat hepatocytes and preferentially stimulates DNA synthesis in γ-glutamyltranspeptidase positive cells. Carcinogenesis, 13: 373-378.
PubMed - Oshiro, Y., P.S. Bawierz and C.E. Piper, 1986. Absence of a genotoxic response from steroids in the rat primary hepatocyte unscheduled DNA synthesis assay. Environ. Mutagen., 8: 461-465.
PubMed - Reimann, R., S. Kalweit and R. Lang, 1996. Studies for a genotoxic potential of some endogenous and exogenous sex steroid. II. Communication: examination for the induction of cytogenetic damage using the chromosomal aberration assay on human lymphocytes in vitro and the mouse bone marrow micronucleus test in vivo. Environ. Mol. Mutagen., 28: 133-144.
PubMed - Roy, D., R.A. Floyd and J.G. Liehr, 1991. Elevated 8-hydroxy deoxyguanosine levels in DNA of diethylstilbestrol treated Syrian hamster: covalent DNA damage by free radicals generated by redox cycling of dimethylstilbestrol. Cancer Res., 51: 3883-3885.
PubMed - Rudali, G., 1975. Induction of tumors in mice with synthetic sex hormones. Gann. Monograph, 17: 243-252.
Direct Link - Schindler, A.E., C. Campagnoli, R. Drucmann, J. Huber, J.R. Pasqualini, K.W. Schweppe and J.H.H. Thijssen, 2003. Classification and Pharmacology of progestins. Maturitas, 46: S7-S16.
PubMed - Sciarra, F., V. Tosano, G. Concolino and F. Di Silverio, 1990. Antiandrogens: Clinical applications. J. Steroid Biochem. Mol. Biol., 37: 349-362.
CrossRefPubMedDirect Link - Siddique, Y.H. and M. Afzal, 2005. Protective role of allicin and L-ascorbic acid against the genotoxic damage induced by chlormadinone acetate in cultured human lymphocytes. Indian J. Exp. Biol., 43: 769-772.
PubMedDirect Link - Siddique, Y.H. and M. Afzal, 2004. Evaluation of genotoxic potential of synthetic progestin chlormadinone acetate. Toxicol. Lett., 153: 221-225.
PubMedDirect Link - Siddique, Y.H. and M. Afzal, 2004. Induction of chromosomal aberrations and sister chromatid exchanges by chlormadinone acetate in human lymphocytes: A possible role of reactive oxygen species. Indian J. Exp. Biol., 42: 1078-1083.
PubMedDirect Link - Siddique, Y.H. and M. Afzal, 2004. Evaluation of genotoxic potential of synthetic progestin ethynodiol diacetate in human lymphocytes in vitro. Curr. Sci., 86: 1161-1165.
Direct Link - Siddique, Y.H. and M. Afzal, 2004. Induction of chromosomal aberrations and sister chromatid exchanges by cyproterone acetate in human lymphocytes. Int. J. Hum. Genet., 4: 187-191.
Direct Link - Siddique, Y.H. and M. Afzal, 2005. Genotoxic potential of cyproterone acetate: A possible role of reactive oxygen species. Toxicol. In vitro, 19: 63-68.
PubMedDirect Link - Siddique, Y.H. and M. Afzal, 2005. In vivo induction of sister chromatid exchanges (SCEs) and chromosomal aberrations (CAs) by lynestrenol. Indian J. Exp. Biol., 43: 291-293.
PubMedDirect Link - Siddique, Y.H. and M. Afzal, 2005. Evaluation of genotoxic potential of norethynodrel in human lymphocytes in vitro. J. Environ. Biol., 26: 387-392.
PubMedDirect Link - Siddique, Y.H., G. Ara, T. Beg and M. Afzal, 2008. Antigenotoxic effect of nordihydroguaiaretic acid against chlormadinone acetate-induced genotoxicity in mice bone-marrow cells. J. Nat. Med., 62: 52-56.
CrossRefDirect Link - Siddique, Y.H., G. Ara, T. Beg and M. Afzal, 2006. Effect of vitamin C on cyproterone acetate induced genotoxic damage in mice. Res. J. Biol. Sci., 1: 69-73.
Direct Link - Siddique, Y.H., G. Ara, T. Beg and M. Afzal, 2006. Genotoxic potential of medroxyprogesterone acetate in culture human peripheral blood lymphocytes. Life Sci., 80: 212-213.
PubMed - Siddique, Y.H., G. Ara, T. Beg, M.H. Shahi and M. Afzal, 2006. Protective role of Ocimum sanctum L. infusion against norethynodrel induced genotoxic damage in cultured human peripheral blood lymphocytes. J. Indian Soc. Toxicol., 2: 10-14.
Direct Link - Siddique, Y.H., T. Beg and M. Afzal, 2006. Protective role of nordihydroguaiaretic acid (NDGA) against norgestrel induced genotoxic damage. Toxicol. In Vitro, 20: 227-233.
Direct Link - Siddique, Y.H., G. Ara, T. Beg and M. Afzal, 2007. Anti-genotoxic effect of Ocimum sanctum L. extract against cyproterone induced genotoxic damage in cultured mammalian cells. Acta Biol. Hung., 58: 397-409.
CrossRefPubMedDirect Link - Siddique, Y.H., G. Ara, T. Beg, M. Faisal, M. Ahmad and M. Afzal, 2008. Antigenotoxic role of Centella asiatica L. extract against cyproterone acetate induced genotoxic damage in cultured human lymphocytes. Toxicol. In vitro, 22: 10-17.
PubMedDirect Link - Siddique, Y.H., T. Beg and M. Afzal, 2005. Antigenotoxic effects of ascorbic acid against megestrol acetate-induced genotoxicity in mice. Hum. Exp. Toxicol., 24: 121-127.
CrossRefPubMedDirect Link - Siddique, Y.H., T. Beg and M. Afzal, 2005. Genotoxic potential of ethinylestradiol in cultured mammalian cells. Chem. Biol. Interact., 151: 133-141.
CrossRefPubMedDirect Link - Siddique, Y.H., T. Beg and M. Afzal, 2006. Protective effect of nordihydroguaiaretic acid (NDGA) against norgestrel induced genotoxic damage. Toxicol. in vitro., 20: 227-233.
CrossRefPubMedDirect Link - Siddique, Y.H., T. Beg and M. Afzal, 2007. Anticlastogenic effects of ascorbic acid against the genotoxic damage induced by norethynodrel. Adv. Environ. Biol., 1: 27-32.
Direct Link - Siddique, Y.H., T. Beg and M. Afzal, 2007. Genotoxic potential of fluoxymesterone in cultured human lymphocytes. Adv. Environ., Biol., 1: 4-10.
Direct Link - Somkuti, S.G., J. Sun, C.W. Yowell, M.A. Fritz and B.A. Lessey, 1996. The effect of oral contraceptive pills on markers of endometrial receptivity. Fertil. Steril., 65: 484-488.
PubMed - Stenchever, M.A., J.A. Jarvis and N.K. Kreger, 1969. Effect of selected estrogens and progestins on human chromosomes in vitro. Obstet Gyneco., 34: 249-251.
PubMed - Topinka, J., B. Binkova, H.K. Zhu, U. Andrae, I. Neumann, L.R. Schwarz, S. Werner and T. Wolff, 1995. DNA damaging activity of cyproterone acetate analogues, chlormadinone acetate and megestrol acetate in rat liver. Carcinogenesis, 16: 1483-1487.
PubMed - Werner, S., S. Kuntz, T. Beckurts, C.D. Heidecke, T. Wolff and L.R. Schwarz, 1997. Formation of DNA adducts by cyproterone acetate and some structural analogues in primary cultures of human hepatocytes. Mutat. Res., 395: 179-187.
PubMed - Wheeler, W.J., L.M. Cherry, T. Downs and T.C. Hsu, 1986. Mitotic inhibition and aneuploidy induction by naturally occurring and synthetic estrogens in Chinese hamster cells in vitro. Mutat. Res., 171: 31-41.
PubMed - Wolf, D.P., L. Blasco, M.A. Khan and M. Litt, 1979. Human cervical mucous V. oral contraceptives and mucus rheologic properties. Fertil. Ster., 32: 166-169.
PubMed - Yager, J.D. and J.G. Liehr, 1996. Molecular mechanisms of estrogens carcinogenesis. Ann. Rev. Pharmacol. Toxicol., 36: 203-232.
PubMed - Zeiger, E., B. Anderson, S. Haworth, T. Lawlor and K. Mortelmans, 1992. Salmonella mutagenicity tests, V. Results from testing 311 chemicals. Environ. Mol. Mutagen, 19: 2-141.
PubMed - Zhu, B.T. and A.H. Conney, 1998. Functional role of estrogen metabolism in target cells: Review and perspectives. Carcinogenesis, 19: 1-27.
PubMed