
Ch. Kalyana Kumar
Department of Genetics, Bhagawan Mahavir Medical Research Centre I C)- I - I
Mahavir Marg, Hyderabad-500004, A.P., India
Sudha Murthy
Indo American Cancer Institute and Research, Centre Road No. 14:
Banjara Hills Hyderabad-500034, A.P., India
Kaiser Jamil
School of Biotechnology (MGNIRSA), University ofMysore, Mysore, India
International Journal of Pharmacology
Year: 2007 | Volume: 3 | Issue: 2 | Page No.: 130-136







ABSTRACT
To determine the frequency of the IVS14+1G>A mutation in the DPD gene in the South Indian population, we have carried out PCR based genotyping allowing the rapid analysis of the IVS14+1G>A mutation by RFLP. For screening for the presence of this mutation a total of 112 breast cancer biopsy samples and 82 healthy controls were included in our study. Out of 112 breast cancer patients 72 individuals were on 5-FU treatment. In this group we identified 6 heterozygous and 2 homozygous mutations confirming the prevalence of about 5.3 and 1.7% mutations in the invasive Ductal carcinomas. In healthy controls out of 82 we found 2 (2.5%) naturally occurring heterozygous mutations. In this study the prevalence of the IVS14+1G>A mutations in IDC’S were found to be significant with an increased risk upon 5-FU administration. Mutations of the DPD gene results in severe DPD deficiency. DPD deficient patients have shown splice-site polymorphism, IVS14+G-A (i.e., a G to A alteration in the nucleotide at the exon14 acceptor splice site), the corresponding mRNA therefore lacks exon 14 and the enzymatic activity of the translated DPD protein being virtually absent. It is therefore concluded that genetic screening for the presence of this mutation in cancer patients would be useful before the administration of 5-FU, in the Indian population suffering from breast cancer.
PDF Abstract XML References
How to cite this article
Ch. Kalyana Kumar, Sudha Murthy and Kaiser Jamil, 2007. Possible Associations of Splice Site Mutation of Dihydropyrimidine Dehydrogenase (IVS14+1G>A) in Adverse Drug Reactions in Some Invasive Ductal Carcinoma Patients. International Journal of Pharmacology, 3: 130-136.
DOI: 10.3923/ijp.2007.130.136
URL: https://scialert.net/abstract/?doi=ijp.2007.130.136
DOI: 10.3923/ijp.2007.130.136
URL: https://scialert.net/abstract/?doi=ijp.2007.130.136
INTRODUCTION
Genetic factors could alter drug metabolism and activity and can predict drug toxicity/or efficacy. Therefore molecular diagnosis is becoming an increasingly important area in chemotherapy of cancers. Pharmacogenetic disorder has been described concerning cancer patients with a complete or partial deficiency of Dihydropyrimidine Dehydrogenase (DPD) who exhibit severe life-threatening toxicity (e.g., diarrhea, stomatitis, mucositis, myelosuppression, granulocytopenia, neurotoxicity and in some cases death) (Van Kuilenburg et al., 2000a-c). DPD is a pyrimidine catabolic enzyme and the initial and rate-limiting step in the pathway of uracil and thymidine catabolism and also in the pathway leading to the formation of beta-Alanine (Van Kuilenburg et al., 2002). The activity of DPD is exclusively present in cytosol and the enzyme seems to be ubiquitously expressed with the highest activity being found in the liver and peripheral blood monocytes (Iyer et al., 1999). Dihydropyrimidine dehydrogenase is a large gene of approximately 150 kb consisting of 23 exons encoding a protein of approximately 111-kDa. It is located on chromosome 1 at position 1p22 (Wei et al., 1998). DPD is also responsible for the breakdown of the widely used antineoplastic drug like 5-fluorouracil (5FU). The catabolic route plays a significant role as more than 80% of the administered 5-fluorouracil is catabolized by DPD (Van Kuilenburg et al., 2000). Even though 5-FU is not prescribed as a single agent, it has been reported to influence the action of other drugs which are given along or subsequently after 5-FU.
5-Fluorouracil (5-FU) remains one of the most frequently prescribed chemotherapeutic drugs for the treatment of cancers of the Breast, Gastrointestinal tract and the Head and neck (Van Kuilenburg, 2004). This molecule was designed to occupy the active sites of enzyme targets, there by blocking metabolism in malignant cells. Although this antimetabolite is toxic, its efficacy makes it one of the most widely used agents against solid tumors. 5-FU is metabolized via two routes in competition with each other: The anabolic route, which gives rise to the active metabolites and the catabolic route, which inactivates 5-FU and leads to elimination of the drug from the system. In the metabolic regulation 5-FU can enter the anabolic pathway that converts it to flurodeoxyuridine monophosphate (FdUMP), which can convert to FluorodeoxyUridinediphosPhate (FdUDP) or can inhibit Thymidylate Synthase (TS). Inhibiting TS prevents the conversion of deoxyuridinemonophoshate to Thymidylate, blocking the proper formation of DNA. FdUDP can converted to flurodeoxyuridine triphosphate, which is added to the growing DNA strands, where it alters the proper functioning of the DNA, leading to cell damage and death (Langenbach et al., 1997; Peters et al., 1995). The catabolic pathway of 5-FU is depicted in Fig. 1, where DPD is shown to play an important role in bioconversions and bioavailability of the drug. Most of the administered dose (85%) of 5-FU is converted by a series of enzymes (of which DPD is the rate limiting step) to inactive metabolites dihydroflurouracil (FUH2), alpha fluoro beta ureidopropiuonate (FUPA) and alpha flourobeta alanine (FBAL).
So far, more than 30 mutations in the DPD gene have been described, at least 20 of which have been reported to be functional and some of them are clearly linked to 5-FU toxicity e.g., IVS14+1G>A point mutation (Van Kuilenburg et al., 2000c). The sequence variations of DPYD gene are represented in Fig. 2.
Subsequently, a number of mutations and polymorphisms have been identified, some of which result in a decreased activity of the DPD enzyme. The most frequent of these mutations is a G to A point mutation within the 5-splicing donor site of intron 14 (exon 14-skipping mutation; DPD splice site mutation). Processing of pre-mRNA bearing this mutation results in a loss of exon 14. The resulting protein product is truncated by 55 amino acids; its catalytic activity is virtually absent (Raida et al., 2001).
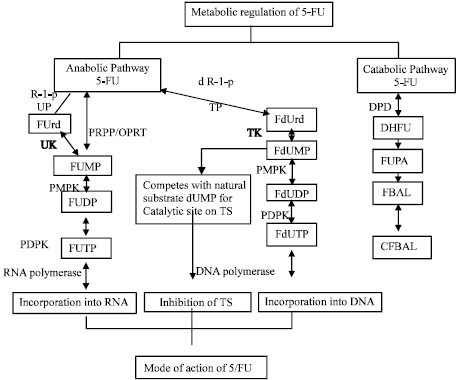 | |
| Fig. 1: | Metabolic Pathway of 5-Fluorouracil (Lee et al., 2004) |
 | |
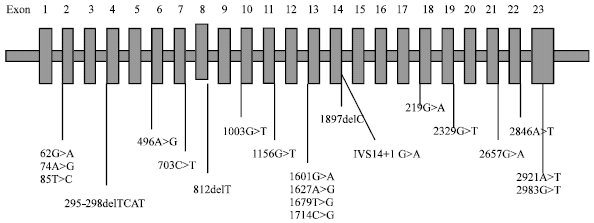 | |
| Fig. 2: | Sequence variations identified in the DPYD gene (Lee et al., 2004) This figure locates the sequence variations identified in the coding and noncoding of the DPYD gene. The location of the positive and negative regulatory elements identified 5’UTR, upstream of the transcription start point is shown on the figure. Other sequence variations that were reported but not shown on the figure include: DPYD*9B, which is composed of both sequence variants DPYD*9A (85T>C; C29R) in exon 2 and 2846A>T (D949V) in exon 23; and DPYD*2B, which is composed of both sequence variants DPYD*2A (IVS14+1G>A) in intron 14 and DPYD*5 (1627A>G; 1543V) in exon 13 |
To date, polymorphisms in genes involved in the synthesis of thymine or uracil have been reported to be associated with the risk of human cancers. For example, genetic polymorphisms of Thymidylate Synthase (TS) and Methylenetetrahydrofolate Reductase (MTHFR) have both been demonstrated to modulate the risk of Breast cancer and other cancers (Kumar and Jamil, 2006; Reddy and Jamil, 2006).
It seems biologically plausible to hypothesize that there could be an association between the gene polymorphisms in DPYD, which catalyze pyrimidine bases and susceptibility to chemotherapy, though no reports on such association have yet been published. Accordingly, in this study, we investigated the associations between DPYD splice-site mutation, IVS14+1G>A (G to A alteration in the nucleotide at the exon14 acceptor splice site) in IDC’S of this region (AP, India) using a prevalent case-control population. In our experiments, we have evaluated the role of DPD polymorphism in patients receiving 5-FU and other drugs. Determination of polymorphisms in drug metabolizing enzymes before the administration of chemotherapy could offer new strategies of optimizing the therapy of individual patients.
MATERIALS AND METHODS
Study population and sample collection: Breast cancer patients were enrolled in this study on the basis of clinical examinations as well as mammography and histopathological examinations Two senior pathologists confirmed all diagnoses. The breast cancer study is a population-based case-control study conducted on South Indian patients. A total of 112 breast cancer patients and 82 age matched controls were enrolled in the study. Written informed consent was obtained from all subjects and relevant ethical committees approved the study
Inclusion and exclusion criteria: All incident breast cancer cases were newly diagnosed during the study period (April 2005 to June 2006) and meeting the following criteria were eligible for this study: 25-68 years age of women, residents of Andhra Pradesh (South India), with no previous history of any cancer. The ethnicity of this group is therefore of Indian origin. The breast cancer patients studied had not been exposed to chemo-and/or radiotherapy before. They underwent clinical examinations at various hospitals in Hyderabad.
Collection of biopsy samples: The specimens obtained were proven cases of breast cancer patients who underwent either breast conservative therapy or radical mastectomy from various institutes like Indo American Cancer Institute and M.N.J. Hospital Hyderabad. The tumor samples obtained were of various tumor sizes and diagnosed mainly as Invasive Ductal Carcinoma or Invasive Lobular Carcinoma.
Collection of blood samples: Blood samples from healthy women were collected by venipuncture. These samples were used as controls.
Genotyping analysis DPD gene IVS14+1G>A mutation: Genomic DNA was extracted from biopsies (tissues) after homogenization of the samples and blood using standard procedure The DPD, IVS14+1G>A Mutations were detected by P.C.R amplification with specific primers, as described below.
PCR amplification of exon 14: PCR amplification of Exon 14 of DPD was adapted as follows. Initial denaturation step was for 4 min at 94°C followed by 30 cycles of denaturation for 30 sec at 94°C, annealing for 30 sec at 56°C and elongation for 90 sec at 72°C. The final elongation step was for 7 min at 72°C. Primers used were: Primer F, 5' TGCAAATATGTGAGGAGGGACC-3 Primer R, CAGCAAAGCAACTGGCAGATT-3 (obtained from Sigma) resulted in a 409-bp product. Each primer was used at 0.25 μmol L-1 in a 25 μL final volume in the presence of standard PCR master mix (Genetrix) amplifications were performed
Restriction fragment length polymorphism of exon14: Amplicons were then digested for at least 2 h (usually 4 h) with MaeII in the buffer supplied by the manufacturer, IVS14+1G>A mutation creates MaeII restriction site causes cleavage of the 409 bp, IVS14+1G>A heterozygotes were identified by 409-, 278- and 131 bp fragments and, IVS14+1G>A homozygotes were identified by 278 and 131 bp fragments. The wild-type allele remains undigested. The products were visualized by Ethidium bromide staining (0.5 mg L-1) in a 4% agarose gel. All the chemicals used were of analytical grade and were obtained from Bioserve and Sigma.
RESULTS
Investigations on DPD IVS14+1G>A mutation in the DPYD gene in the Breast Cancer patients and in healthy controls was carried out and results presented in a table and gel picture. Specific primers were used for the amplification of exon 14 and its flanking intron sequence. Present analysis showed that Breast Cancer patients were heterozygous and homozygous for IVS14+1G>A mutation which changes invariant splice site G-A.
Figure 3 showing agarose gel electrophoresis for the detection of mutant DPYD alleles. Lane M: 100 bp Marker. Lane 1 and 2 homozygous mutant subjects, lane 3 wild type and lane 4 and 5 heterozygous mutant subjects.
It was seen that the wild type allele gives 409 bp band, heterozygous mutant allele gives 278 and 131 bp bands and homozygous mutant allele gives 409, 238 and 131 bp bands.
The frequency distribution through analysis of 112 breast cancer patients and 82 healthy controls for the presence of the IVS14+1G>A mutation showed 6 patients with heterozygous and 2 homozygous mutations in breast cancer patients and 2 heterozygous mutations in controls (Table 1). The statistical analysis of the frequency of heterozygous was 5.3% and the frequency of homozygous was 1.7% in breast cancer patients and 2.5% in controls (Table 1). These results showed the prevalence of the IVS14+1G>A mutation in breast cancer patients, hence it is possible that these patients may have increased risk to experience toxicity effect upon 5-FU administration.
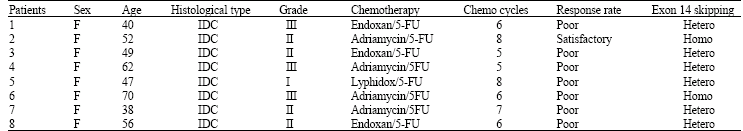
Table 2 represents only those cases with DPD polymorphism (G>A). It is seen from these results that women between the age group of 38-70 years have shown poor response to 5-FU. Also we found that most of these patients were diagnosed with Grade II and III tumors. After surgery we followed their cases and found that these were given a combination drugs like Endoxan+5-FU and Adriamycin+5FU and the cases with the splice site polymorphisms were found to be poor responders.
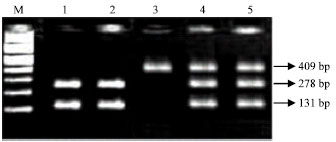 | |
| Fig. 3: | RFLP (MaeII) Gel Picture |
| Table 1: | The genotype frequencies of G>A mutations in the breast cancer patients and controls |
 | |
| Table 2: | Characteristic and DPD Exon 14-skipping allelotype of breast cancer patients with 5-FU based chemotherapy |
 | |
DISCUSSION
Adverse drug reactions are a great concern to both healthcare practitioners and the general public, accounting for over 100,000 fatalities per year; it is the fourth leading cause of death in the United States, after heart disease, cancer and stroke. It is now clear that genetic differences in drug metabolizing enzymes, transporters, receptors may result in altered drug response including both efficacy and toxicity. The small difference between the mean efficacious dose and the mean toxic dose make chemotherapeutic agent’s prime candidates for the occurrence of interindividual variability in drug efficacy and toxicity. A first report of a heterozygous carrier of the exon 14-skipping mutation who had experienced severe toxicity after 5-FU administration was published in 1996. Subsequently, additional cases have been reported. On the basis of these reports it was clear that screening of cancer patients routinely for the exon 14-skipping mutation before 5-FU treatment would be useful (Johnson et al., 1999). The aim of this investigation has also been to examine the polymorphism with respect to ethnicity. Recently an in vitro study has reported that certain drugs require higher concentrations to induce the toxic effects. Hence in such cases larger doses of certain drugs are required to bring about the desired effect (Suman and Jamil, 2006). Pharmacogenetic studies on chemotherapeutic agents have the potential to revolutionize the way clinicians determine a patient’s optimal treatment regimen through the prediction of an individual’s responsiveness to a particular therapy.
Analysis of the prevalence of the various mutations among cancer patients showed that the G>A point mutation in the invariant splice donor site is the most common one (43%) (Van Kuilenburg et al., 1997). This observation is the line with the fact that the IVS14+1G-A mutation is most predominant mutation detected in cancer patients. G to A mutation in the 5’-splicing consensus sequence in the human DPYD gene was uncovered that leads to skipping of the entire exon preceding the mutation (Van Kuilenburg et al., 1997). In the 5’splicing consensus sequence, the dinucleotide GT is the best conserved among mammals and mutations affecting this sequence were reported to cause the inactivation of several genes associated with human diseases including phenylalanine hydroxylase, procollagen COL3A1 and hypoxanthine guanine phosphoribosi-transferase. In these genes, a G to A mutation in the GT splicing recognition sequence consistently results in skipping of the entire exon preceding the mutation (Wei et al., 1996).
A point mutation (G to A) within the GT 5-splicing consensus sequence of exon 14 of the human DPD gene has been shown to result in the skipping of the entire exon 14. This exon-skipping causes a deletion of 55 amino acid residues in the primary sequence of the DPD protein resulting in a lack of functional DPD expression (Meinsma et al., 1995; Verken et al., 1996). The exon 14-skipping mutation is the most abundant cause for the loss-of-function allele of the DPD gene known thus far (Van Kuilenburg et al., 1999a).
Polymorphism in various ethnic groups: Several reports have been made on the prevalence of the exon 14-skipping mutation in different ethnic population (Table 3). However, the sample size in all of these reports was too small to yield reliable and consistent data, thus leading to conflicting conclusions. Carriers of the mutant allele were reported in healthy individuals of the following cohorts studied: Finnish (2 of 45 heterozygotes); Taiwanese (2 of 36 heterozygotes; (Meinsma et al., 1995) and Finnish (2 of 90 heterozygotes; Van Kuilenburg et al., 1999b). In addition, one heterozygote was detected in a sample of 72 colorectal cancer patients (Wei et al., 1998.). In other small cohorts of healthy donors, however, no carrier of the mutant allele was detected: British (30 individuals; Meinsma et al., 1995); Taiwanese (131 individuals); Japanese (90 individuals); African-American (105 individuals); (Wei et al., 1996) British (30 with low and 30 with high DPD catalytic activity in blood mononuclear cells (Ridge et al., 1998); and Dutch (50 individuals; (Verken et al., 1996) ). It was estimated that 3% of the normal population could be heterozygous for the DPYD allele and using the Hardy-Weinberg equilibrium upto 1 in every 1000 births in the general population could be homozygous of DPYD mutation.
Present data shows that the mutation occurs with a frequency of 5.3% in heterozygous and 1.7% in homozygous within the Indian breast cancer patients and 2.5% heterozygous mutations were detected in controls. We detected this mutation in a heterozygous allelic state in 6 out of 112, all the 6 patients were receiving 5-FU and the response rate was found to be poor and 2 of 112 homozygous mutants were detected in breast cancer patients these 2 patients were also on 5FU therapy and the response rate was poor.
| Table 3: | Different ethnic prevalence of IVS14+1G-A polymorphism |
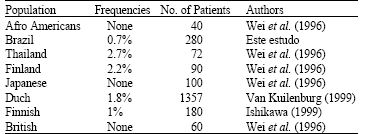 | |
In healthy control we identified 2 heterozygous mutations. In our studies we observed that the breast cancer patients had shown prevalence of G to A point mutation in the invariant splice donor site. This mutation leads to skipping of exon 14 immediately upstream of the mutated splice donor site in the process of DPD pre-mRNA splicing. As a result, the mature DPD mRNA lacks a 165 segment encoding the amino acid 581-635. Therefore it is possible that in our studies the DPD IVS14+1G>A polymorphism is associated with risk of toxicity with 5FU in breast cancer women. Previous studies pointed out that not everybody who is heterozygous for the mutation may actually suffer severe 5-FU side effects. The individuals with heterozygous mutations were found to suffer from toxic life-threatening 5-FU-related toxicity can only be addressed by pharmacokinetic measurements of the individual 5-FU turnover in heterozygous patients using very low doses of 5-FU. This exon 14 splice site mutation has also been suggested to be a common mechanism for DPD deficiency and severe 5-FU toxicity in patients with cancer (Van Kuilenburg et al., 1997). Even incomplete deficiency of DPD is sufficient to cause severe toxicity with 5-FU. Since 5FU is the most commonly prescribed chemotherapeutic drug in cancer treatment and the mutant DPYD allele reported here appears to be homogenized among different ethnic groups, the screening of cancer patients a priori could be feasible approach for avoiding 5FU toxic effects. It can also be used for the determination of mutation carriers and for prenatal diagnosis of DPD deficiency.
CONCLUSIONS
Genotyping enhances precision regarding the choice of drug, or the dose of the drug, in instances when the medication has a narrow therapeutic dose range, when the consequences of treatment failure are severe and/or when critical adverse reactions are more likely in patients with certain polymorphisms. Detecting the DPYD IVS14+1G>A mutation is important information. Even though this is a small study the information generated may be important in decision making about the drug interventions in cancer patients.
REFERENCES
- Johnson, M.R., A. Hageboutros, K. Wang, L. High, J.B. Smith and R.B. Diasio, 1999. Life-threatening toxicity in a dihydropyrimidine dehydrogenase-deficient patient after treatment with topical 5-fluorouracil. Clin. Cancer Res., 5: 2006-2011.
Direct Link - Lee, A., H. Ezzeldin, J. Fourie and R. Diasio, 2004. Dihydropyrimidine dehydrogenase deficiency: Impact of pharmacogenetics on 5-Fluorouracil therapy. Clin. Adv. Hematol. Oncol., 2: 527-532.
Direct Link - Meinsma, R., P. Fernandez-Salguero, A.B.P van Kuilenburg, A.H van Gennip and F.J. Gonzalez, 1995. Human polymorphism in drug metabolism: Mutation in the dihydropyrimidine dehydrogenase gene results in exon skipping and thymine uraciluria. DNA Cell Biol., 14: 1-6.
Direct Link - Raida, M., W. Schwabe, P. Hausler, A.B. Van Kuilenburg, A.H. Van Gennip, D. Behnke and K. Hoff Ken, 2001. Prevalence of common point mutation in the Dihydropyrimidine Dehydrogenase (DPD) gene with in the 5'-Splice donor site of intron 14 in patients with severe 5-fluorouracil(5FU) related toxicity compared with controls. Clin. Cancer Res., 7: 2832-2839.
Direct Link - Reddy, H. and K. Jamil, 2006. Polymorphism in the MTHFR gene and their possible association with susceptibility to childhood acute lymphocytic leukemia in an Indian population. Leukemia Lymphoma, 47: 1333-1339.
Direct Link - Ridge, S.A., J. Sludden, O. Brown, L. Robertson and X. Wei et al., 1998. Dihydropyrimidine dehydrogenase pharmacogenetics in caucasian subjects. Br. J. Clin. Pharmacol., 46: 151-156.
Direct Link - Suman, G. and K. Jamil, 2006. Application of human lymphocytes for evaluating toxicity of anti-cancer drugs. Int. J. Pharmacol., 2: 374-381.
CrossRefDirect Link - Van Kuilenburg, A.B.P., P. Vreken, L.V. Beex, R. Meinsma and H. Van Lenthe et al., 1997. Heterozygocity for a point mutation in an invariant splice donor site of dihydropyrimidine dehydrogenase and severe 5-fluorouracil related toxicity. Eur. J. Cancer, 33: 2258-2264.
Direct Link - Van Kuilenburg, A.B.P., H. Van Lenthe, M.J. Blom, E.P.J. Mul and A.H. Van Gennip, 1999. Profound variation in dihydropyrimidine dehydrogenase activity in human blood cells: Major implication Br. J. Cancer, 79: 620-626.
Direct Link - Van Kuilenburg, A.B.P., P. Vreken, N.G.G. Abeling, H.D. Bakker and R. Meinsma et al., 1999. Genotype and phenotype in patients with dihydropyrimidine dehydrogenase deficiency. Hum. Genet., 104: 1-9.
Direct Link - Van Kuilenburg, A.B.P., E.W. Muller, J. Haasjes, R. Meinsma and L. Zoetekouw et al., 2000. Lethal outcome of a patient with a complete Dihydropyrimidine Dehydrogenase (DPD) deficiency after administration of 5-Fluorouracil: Frequency of the common IVS14 1G"A mutation causing DPD deficiency. Clin. Cancer, 1: 1149-1153.
Direct Link - Van Kuilenburg, A.B.P., J. Haasjes, D.J. Richel, L. Zoetekouw and H. Van Lenthe et al., 2000. Clinical implications of Dihydropyrimidine Dehydrogenase (DPD) deficiency in patients with severe 5-Fluorouracil-associated toxicity: Identification of new mutations in the DPD gene. Clin. Cancer Res., 6: 4705-4712.
Direct Link - Van Kuilenburg, A.P.B., A.H. Tromp, P.C.J. Valtman and A.H. Van Gennip, 2000. Pitlls in diagnosis of patients with a partial dihydropyrimidine dehydrogenase deficiency. Clin. Chem., 46: 9-17.
Direct Link - Van Kuilenburg, A.B., 2004. Dihydropyrimidine dehydrogenase and the efficacy and toxicity of 5-fluorouracil. Eur. J. Cancer, 40: 939-950.
Direct Link - Wei, X., H.L. McLeod, J. McMurrough, F.J. Gonzalez and P. Fernandez-Salguero, 1996. Molecular basis of the human dihydropyrimidine dehydrogenase deficiency and 5-fluorouracil toxicity. J. Clin. Investigat., 98: 610-615.
Direct Link - Wei, X., G. Eizondo, A. Sapone, H.L. McLeod and H. Raunio et al., 1998. Characterization of the human dihydropyrimidine dehydrogenase gene. Genomics, 51: 391-400.
CrossRef