
Enas M. Moustafa
Department of Radiation Biology, National Center for Radiation Research and Technology, Atomic Energy Authority, Cairo, Egypt
LiveDNA: 20.16969
Noura M. Thabet
Department of Radiation Biology, National Center for Radiation Research and Technology, Atomic Energy Authority, Cairo, Egypt
Khaled Sh. Azab
Department of Radiation Biology, National Center for Radiation Research and Technology, Atomic Energy Authority, Cairo, Egypt
International Journal of Cancer Research
Year: 2017 | Volume: 13 | Issue: 2 | Page No.: 41-50







ABSTRACT
Background and Objective: Atorvastatin (AV) is statin drug that lower peripheral cholesterol production to prevent cardiovascular disease and became of interest in cancer prevention. This study was to articulate the possible inhibitory potential of AV and gamma radiation on STAT-3 and β-catenin signals during cancer evolution. Materials and Methods: Ehrlich ascites carcinoma (EAC) cells (2.5×106 cells/mouse) were inoculated to mice subcutaneously in the right thigh at zero time synergistically with starting AV administration (10 mg kg–1 b.wt.,/day for 21 days). Accordingly, mice were exposed to (4 Gy) of gamma radiation at the 13th days from experiment commence. The statistical analysis of the results was performed by one-way analysis of variance (ANOVA). Results: MTT assessment identified that AV addition to culture of EAC cells in a proper concentrations decreased cell viability in a dose dependent manner: (IC50 = 512 mg L–1: LD50 = 63 mg kg–1). The injection of the AV and the exposure to gamma radiation markedly decreased the tumor volume when compared to E mice. Further, the concentrations of lipid peroxides (MDA) and Glutathione (GSH) as well as the activities of glutathione peroxidase (GPX) and glutathione transferase (GST) were significantly ameliorated, in addition to decreased in the levels of β-catenin and STAT-3 mRNA expression and the concentration of p-STAT-3. Also, the expression of p53 mRNA is significantly improved when AV administrated. Conclusion: Credibly, the AV blockade of STAT-3 convenient activation might turned out the β-catenin expression, leading to stopping tumor progression. Exposure of mice bearing Ehrlich solid tumor to gamma radiation seemed to have a capability to proceed the action of AV. Actually, the overall results support the antitumor effect of AV which could be potentiated by radiotherapy.
PDF Abstract XML References Citation
Received: August 21, 2017;
Accepted: September 15, 2017;
Published: September 25, 2017
How to cite this article
Enas M. Moustafa, Noura M. Thabet and Khaled Sh. Azab, 2017. Statin Alter Expression of STAT-3 and ß-Catenin Signal Molecules in Gamma Irradiated Model of Carcinoma. International Journal of Cancer Research, 13: 41-50.
DOI: 10.3923/ijcr.2017.41.50
URL: https://scialert.net/abstract/?doi=ijcr.2017.41.50
DOI: 10.3923/ijcr.2017.41.50
URL: https://scialert.net/abstract/?doi=ijcr.2017.41.50
INTRODUCTION
Statins, the 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, are a class of drugs that inhibit the rate-limiting step in the cholesterol biosynthetic pathway1. Besides their use in the treatment of lipid disorders, statins have been shown anti-proliferative and pro-apoptotic effects in malignant2. In addition, substantial experimental and clinical evidence suggests that statins exhibit anticancer effects mediated by apoptosis and cell cycle arrest through various signaling pathways3.
Signal transducers and activators of transcription (STAT) is a family of transcription factors. STAT-3 is one of six members of this family that has been associated with inflammation, survival, proliferation, angiogenesis, metastasis and chemo resistance of tumor cells4. Activation of STAT-3 occurs following ligand binding to a variety of membranous receptors including receptors for cytokines, hormones and growth factors. However, various intracellular kinases, such as the SRC family kinases, can activate STAT-3 without receptor involvement. Activated STAT-3 is usually phosphorylated on Tyrosine 705 and in this form, it translocates to the nucleus where it can act as a transcription factor. Amongst its targets are molecules involved in cell proliferation [such as Cyclin D1 and C-Myc], apoptosis [such as BCL-2 and BCL-X] and cell motility [such as TWIST]5. Apparently, this factor constitutively expressed in multiple myeloma (MM), leukemia, lymphoma, squamous cell carcinoma and other solid tumors, including cancers of the prostate and breast6. As a matter of fact, Beta-catenin (β-catenin), a nuclear and cytoplasmic expressed protein, is a vital component of the canonical Wnt/β-catenin signaling pathway which is described as an oncogenic cause in many human cancers. In cytoplasm, β-catenin degraded by the action of destruction complex which assembled in the absence of Wnt signaling. However, in the presence of Wnt signaling, β-catenin destruction complex disassembled resulting in β-catenin accumulation that enters the nucleus to initiate its oncogenic function. Accordingly, β-catenin inhibitors in combination with standard systemic therapies holds great promise to improve treatment’s efficacy and outcome7.
Ehrlich ascites carcinoma (EAC) is actually one of the commonest experimental tumors exploit for chemotherapeutic studies. It is a spontaneous murine mammary adenocarcinoma adapted to ascites form. EAC has high transplantable capability, no regression, rapid proliferation, shorter life span, 100% malignancy and that also does not have tumor-specific transplantation antigen (TSTA). EAC could be used as ascites (cells injected intra-peritoneally, i.p.) or a solid form (cells injected subcutaneously, s.c.)8-10.
As, gene products like STAT-3 or β-catenin are closely related to tumor development and growth, agents that can inhibit the expression of STAT-3 and β-catenin may have great potential in the treatment of cancer and other inflammatory diseases. Thus, agents that disrupt these pathways would be good candidates for STAT-3 inhibitors. So as it is shown, this study aimed to investigate the effect of atorvastatin and gamma irradiation on the interactions of STAT-3 and β-catenin during progression of Ehrlich solid tumor in mice.
MATERIALS AND METHODS
Animals categorize: In this experimental study, adult female Swiss albino mice weighing (22-25 g) were obtained from the breeding unit of the Egyptian Organization for Biological Products and Vaccines (Cairo). The animals were acclimatized for one week and maintained on a commercial standard pellet diet and water ad libitum. Animal experimentation was conducted under national research centre guidelines for the use and care for laboratory animals and were approved by an independent ethics committee of National Center for Radiation Research and Technology, Atomic Energy Authority, Cairo, Egypt. All of the study procedures have taken place during the year 2014 . In this study, it was used 100 female albino mice weighing about 22-25 g. The animals were categorized into 5 equal groups, 20 mice of each as follows: Control: Mock mice: Ehrlich (E), mice injected in thigh with EAC cells, zero time of experiment: E+AV, mice injected with EAC cells and received AV for 21 days started at zero time of experiment: E+R, mice injected with EAC cells and exposed to 4 Gy on the 13th day from the experiment commence, E+AV+R: Mice injected with EAC cells, received AV and exposed to 4 Gy. Mice were sacrificed on 14th and 21st of experiment. The skeletal muscle (normal control) and tumor tissues were collected for biochemical investigation.
Materials: Atorvastatin (AV) was obtained from (Lipitor®, Pfizer Australia Pty Ltd, Pfizer New Zealand Ltd Auckland, New Zealand). Other chemicals and reagent in this study were obtained from Sigma-Aldrich Chemical Co., USA.
Ehrlich solid carcinoma tumor model: Because a model of Ehrlich Solid Tumor (EST) was used, Ehrlich Ascites Carcinoma cells (EAC) obtained from the Pharmacology and Experimental Oncology Unit of the NCI, Cairo University. EAC cells originated from human breast cancer which modified to grow in female Swiss albino mice and maintained by i.p. of 2.5 million carcinoma cells in the mice. The EAC cells were counted before i.p., using the bright line haemocytometer and dilution was done using physiological sterile saline solution. So, to asses EST in thigh, 0.2 mL EAC cells (2.5×106 cells/mouse) were inoculated (s.c.) in the right thigh of the lower limb of female mouse9-11.
Assay for in vitro cytotoxicity: In vitro short term, cytotoxicity of AV was assayed using EAC cell line. Briefly, 1×106 EAC cells suspended in 100 μL of phosphate buffered saline (PBS, 0.2 M, pH = 7.4) were mixed with 100 μL of various concentrations (1, 50, 200, 400, 800, 1200, 2400 and 3200 μg) of AV and final volume was adjusted to 1 mL with PBS. The mixture was incubated at 37°C for 30 min. Meanwhile, cell suspension in PBS without AV served as control. After the incubation, the viability of the cells was determined, using trypan blue (0.4% in normal saline) dye as per Dolai et al.12. Then, the percentage of cytotoxicity was determined by calculating % inhibition and IC50 values of AV.
Accordingly, to obtain a model for the prediction of lethal dose LD50 values from IC50 values, as reported previously13, a linear regression, from the log transformed IC50 (μg mL–1) and log transformed rodent (μg mL–1) values, were calculated. Then, the equation, which was obtained from a correlation between values of in vitro cytotoxicity and the animal LD50 values for chemicals, was:
| (1) |
Dose of atorvastatin: Thus, administration of AV at dose 10 mg kg–1 b.wt., a pre-standardized and well tolerated dose for in vivo studies to mice was calculated based on a published study that mimics the amount of AV in the range of pharmacological dose for human14.
Irradiation route: The exposure of mice to whole body γ-irradiation was performed with a Canadian gamma cell-40, (137Cs) at the National Center for Radiation Research and Technology (NCRRT), Cairo, Egypt at a dose rate of 0.46 Gy min–1.
Biochemical assay: Lipid peroxidation in tumor tissue was measured by thiobarbituric acid assay, which is based on malondialdehyde (MDA) reaction with thiobarbituric acid, forming thiobarbituric acid reactive substances (TBARS), a pink colored complex exhibiting a maximum absorption at 532 nm accordingly15. The reduced glutathione (GSH) content according to the method of Ellman16. So, the glutathione peroxidase (GPX) activity was assayed according to the method of Gross et al.17 and the glutathione-S-transferase (GST) activity was assayed according to method of Habig et al.18. And as, The conjugation of 1-chloro-2, 4-dinitrobenzene (CDNB) with GSH is accompanied by an increase in absorbance at 340 nm, the rate of increase is directly proportional to the GST activity in the sample.
Quantitative real-time PCR
RNA isolation and reverse transcription: RNA was extracted from the tumor tissue homogenate using the RNeasy plus mini kit (Qiagen, Venlo, The Netherlands), according to the manufacturer’s instructions. Genomic DNA was eliminated by a DNase on column treatment supplied with the kit. The RNA concentration was determined spectrophotometrically at 260 nm, using the NanoDrop ND-1000 spectrophotometer (Thermo Fisher scientific, Waltham, USA) and RNA purity was checked by means of the absorbance ratio at 260/280 nm. Additionally, RNA integrity was assessed by electrophoresis on 2% agarose gels. Therefore, RNA (1 μg) were used in the subsequent cDNA synthesis reaction, which was performed using the Reverse Transcription System (Promega, Leiden, The Netherlands). And total RNA was incubated at 70°C for 10 min to prevent secondary structures. The RNA was supplemented with MgCl2 (25 mM), RTase buffer (10X), dNTP mixture (10 mM), oligo (dt) primers, RNase inhibitor (20 U) and AMV reverse transcriptase (20 U μL–1). This mixture was incubated at 42°C for 1 h. Therefore, quantitative real time PCR: qRT-PCR was performed in an optical 96-well plate with an ABI PRISM 7500 fast sequence detection system (Applied Bio-systems, Carlsbad, California) and universal cycling conditions of 40 cycles of 15 sec at 95°C and 60 sec at 60°C after an initial denaturation step at 95°C for 10 min. Each 10 μL reaction contained 5 μL SYBR Green Master Mix (Applied Bio-systems), 0.3 μL gene-specific forward and reverses primers (10 μM), 2.5 μL cDNA and 1.9 μL nuclease-free water. The sequences of PCR primer pairs used for each gene are shown in Table 1. Later, data were analyzed with the ABI prism sequence detection system software and quantified, using the v1·7 sequence detection software from (PE Biosystem, Foster City, CA, USA). Relative expression of studied genes was calculated by the comparative threshold cycle method. All values were normalized to the endogenous control GAPDH19.
| Table 1: | Primers used for qRT-PCR |
 | |
Western blot analysis of p-STAT-3: In fact, Part of tissue was homogenized with RIPA buffer containing 5 M NaCl, 1 mM phenylmethylsulfonyl fluoride (PMSF), 10% deoxycholic acid (DOC), 10% SDS, 1 M Tris (pH 8.6). The tissue lysate was centrifuged at 12000 rpm for 20 min at 4°C. The lysate was then collected and the protein concentration was determined with a BCA protein assay kit (Thermo Fisher Scientific Inc., USA). An aliquot of 7.5 μg protein of each sample was denatured, then each sample was loaded on 8% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (PAGE) and transferred to a nitrocellulose membrane (Amersham Bioscience, Piscataway, NJ, USA), using a semidry transfer apparatus (Bio-Rad, Hercules, CA, USA). The membranes were incubated with 5% milk blocking buffer containing 10 mM Tris-HCl (pH 7.4), 150 mM NaCl and Tris-buffered saline with 0.05% Tween-20 (TBST) at 4°C overnight. The membranes were then washed with TBST and incubated with a 1:1,000 dilution of anti-STAT-3 (phosphorylated) (Thermo Fisher Scientific Inc., USA) for overnight on a roller shaker at 4°C. For that purpose, the filters were washed and subsequently probed with horseradish peroxidase conjugated anti-mouse immunoglobulin (Amersham. Life Science Inc., USA). Chemiluminescence detection was performed with the Amersham detection kit, according to the manufacturer’s protocols and exposed to X-ray film. The amount of studied protein was quantified by dens isometric analysis of the autoradiograms using a scanning laser densitometer (Biomed Instrument Inc., USA). Results were expressed after normalization for β-actin protein expression (as housekeeping protein)20.
Tumor volume monitoring: Initially, tumors growth analysis, the mice were gauged by vernier caliper on the 14th and the 21st days during the experimental period. The volume of solid tumor was calculated, using formula [A×B2×0:52], where A and B are the longest and the shortest diameter of tumor, respectively21.
Statistical analysis: Statistical analysis was performed by one-way analysis of variance (ANOVA) followed by Duncan’s multiple range test, using statistical package of social science (SPSS) version 15.0 for windows (SPSS Inc., Chicago, IL, USA).All data were presented as the Mean±standard error (SE) p<0.05 were considered as level of significance.
RESULTS
In vitro cytotoxicity assay: AV,at different concentration (1, 50, 200, 400, 800, 1200, 2400 and 3200 μg mL–1), was incubated with EAC for 48 h. The data represented in (Fig. 1 ) display that the IC50 dose of AV was 512 mg L–1: The most antitumor effect showed by significant decreasing in the viability of EAC cells (Fig. 1). In addition, LD50 value for mice was calculated by log IC50 value substitution in Eq. 1 and the result was that the implicated AV dose (10 mg kg–1 day–1) was equivalent to approximately 1/6 of the experimental LD50.
Influences of AV on MDA, GSH Levels, GPX and GST activities: MDA concentration in thigh muscles of control mice was 111.14±2.05 mole g–1 wet tissue in 14th day and 106.27±2.17 mole g–1 wet tissue in 21st day (Table 2). In tumor tissue of E group, significant elevation was resulted in MDA concentration compared to control group. Furthermore, exposure to γ-radiation display significant p<0.05 decrease compared to E group on the 21st experimental intervals. Also, AV treatment to E+AV and E+R+AV groups revealed significant p<0.05 reduction in MDA content on both interval days compared to E group (Table 2)
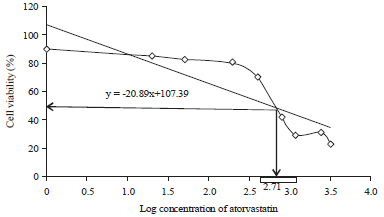 | |
| Fig. 1: | Sigmoidal curve, anti-proliferative effect of AV against EAC cells and represented IC50 value |
| Table 2: | Influences of AV on MDA, GSH levels, GPX and GST activities in tumor tissues |
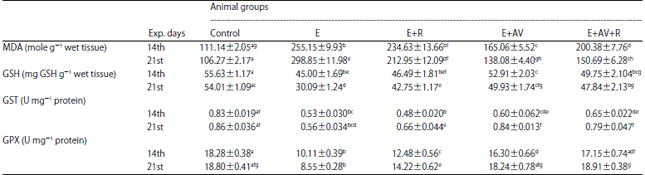 | |
Each value represents the Mean±SE (n = 10). Values with different superscripts are significantly different, at p<0.05 and values with same superscripts not-significantly different, at p<0.05. Control: Mock mice, E: Mice injected with EAC cells, E+AV: Mice injected with EAC cells and received AV, E+R: Mice injected with EAC cells and exposed to 4 Gy, E+AV+R: Mice injected with EAC cells, received AV and exposed to 4 Gy | |
The reduced GSH levels were 55.63±1.17 and 54.01±1.09 mg GSH g–1 weight tissue in control mice on the 14th and the 21st day, respectively (Table 2). In E group, the data showed significant p<0.05 decrease in GSH levels on the 2 experimental intervals compared to control mice. In E+R group, significant p<0.05 increase showed in GSH level on 21st day compared to E group. Surprisingly, Administration of AV to E and E+R groups display significant amelioration in these levels compared to E group. As a result, the activity of GST was 0.83±0.019 and 0.86±0.036 U mg–1 protein in control mice on 14th and 21st day, respectively (Table 2). In EAC bearing mice, the GST activity revealed notable decrease compared to controlling mice on both interval days. On the other hand, In E+R group, the activity of enzyme showed no significant change compared to E group on 14th and significant increase on 21st day. In E+AV and E+AV+R groups, AV treatment showed substantial increase in GST activity compared to E group on both interval days of experiment (Table 2).
In Table 2, GPX activity in control, mice was 18.28±0.38 and 18.80±0.41 U mg–1 protein, respectively on the 14th and the 21st day. In EAC bearing mice, GPX activity demonstrated significant p<0.05 reduction compared to control mice on both interval days. Exposure to γ, radiation showed significant decrease on 14th day and attenuate on 21st day compared to E group. Treatment with AV display noteworthy elevation in GPX activity in both group E and E+R as compared to E group on the 2 experimental intervals.
Influences of AV on STAT-3, p-STAT-3, β-catenin and suppressed tumor gene p53: The data obtained revealed that STAT-3 mRNA of control mice was 1.02±0.019 on the 14th day and was 1.01±0.021 on the 21st day of time interval (Fig. 2a). Also, the protein expression of p-STAT-3 control mice was (1.02±0.021) and (1.00±0.022) on the 14th and the 21st day, respectively (Fig. 2b-c). Meanwhile, β-Catenin mRNA of control mice was 1.06±0.022 on the 14th day and 1.03±0.022 on the 21st day (Fig. 2d).
The results in E group showed a significant p<0.05 increase in expression of STAT-3 mRNA, p-STAT-3 protein expression and β-catenin mRNA compared to control mice in both days (Fig. 2).
Exposure to γ-radiation significantly p<0.05 decrease STAT-3 mRNA, p-STAT-3 protein expression and β-catenin mRNA compared to E group (Fig. 2). Administration of AV induced significant ameliorate in STAT-3 mRNA, p-STAT-3 protein expression and β-catenin mRNA of E+AV and E+AV+R groups compared to E group (Fig. 2).
In addition, the expression of tumor suppressor oncogene (p53 mRNA) in control mice was 1.10±0.0022 on 14th day and 1.03±0.023 on 21st day of time interval (Fig. 2e). The data in E group revealed significant p<0.05 decrease in both interval days compared to control mice (Fig. 2e). In E+R group, significant increase in p53 mRNA expression in 2 experimental days compared to E group. In E and E+R group administrated with AV display significant improvement in both intervals days compared to E group.
Influences of AV on solid tumor volume: The tumor growth manifested by change in tumor volume revealed that, in solid tumor, the tumor volume of E-group was (48.48±1.76 mm3) on 14th and (91.67±3.67 mm3) on 21st (Fig. 3). In E+R group (Fig. 3), no remarkable change was revealed on the14th but rather significant p<0.05 decrease in tumor growth on 21st was observed compared to E group. In E+AV and E+R+AV, administration of AV displayed on the 14th and the 21st significant p<0.05 decreases in tumor growth compared to E group (Fig. 3).
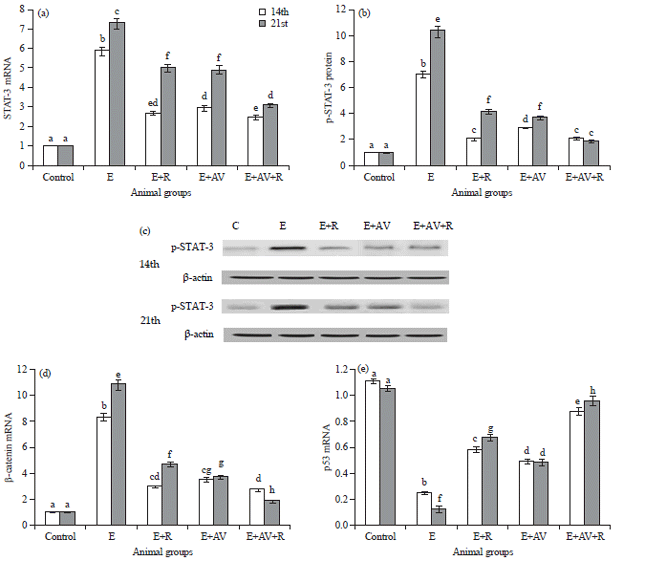 | |
| Fig. 2(a-e): | Influences of AV on (a) Gene expression of STAT-3 mRNA (real time-PCR), (b) Protein expression of p-STAT-3 (Western blot analysis), (c) PAGE of p-STAT-3 (92 kDa) proteins and β-actin (42 kDa) as housekeeping protein, (d) Gene expression of β-catenin mRNA and (e) Gene expression of p53 mRNA |
Each value represents the Mean±SE (n = 10). Values with different superscripts are significantly different, at p<0.05 and values with same superscripts not-significantly different, at p<0.05. Control: Mock mice, E: Mice injected with EAC cells, E+AV: Mice injected with EAC cells and received AV, E+R: Mice injected with EAC cells and exposed to 4 Gy, E+AV+R: Mice injected with EAC cells, received AV and exposed to 4 Gy | |
DISCUSSION
The improper balance between reactive oxygen metabolites (ROMs) and antioxidant defenses results in "oxidative stress", which deregulates the cellular functions leading to various pathological conditions. Overproduction of ROMs induced by different exogenous and endogenous mechanism may exhaust the antioxidant systems of cells and contribute to a number of destructive. Epidemiological studies have suggested that high endogenous level of oxidative adducts and deficiencies in antioxidant levels are likely to be important risk factors for cancer22.
Herein, the data obtained reveals a significant elevation in MDA concentration accompanied with marked inhibition in GPX and GST activities as well as in GSH concentration in solid tumors bearing mice (E) at the 2 experimental intervals compared to normal control (Table 2). Present results were supported by the finding of Pandya et al.22, Metwally et al.23 and Ali et al.24. This results could be attributed to the disturbances occurred in cellular antioxidant system during cancer transformation which leads to accumulation of lipid peroxides (MDA). The end product of lipid peroxidation (MDA) is an autocatalytic free radical chain propagating reaction and known to be associated with pathological conditions of a cell22. Ali et al.24, stated that tumor growth disrupts the antioxidant system and increases lipid peroxidation in tumor host vital organs of E-bearing mice.
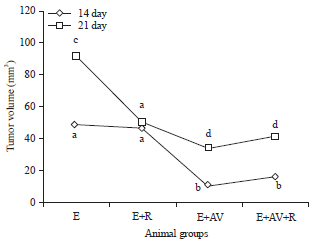 | |
| Fig. 3: | Influence of AV on tumor volume |
Each value represents the Mean±SE (n = 6). Values with different superscripts significantly different, at p<0.05 and values with same superscripts not-significantly different, at p<0.05. Control: Mock mice, E: Mice injected with EAC cells, E+AV: Mice injected with EAC cells and received AV, E+R: Mice injected with EAC cells and exposed to 4 Gy, E+AV+R: Mice injected with EAC cells, received AV and exposed to 4 Gy | |
Collimated reduction in GPX, GST activities as well as the GSH concentration could be attributed to the loss of exact redox tone in EAC bearing mice. In EAC solid tumor, the ratio of GSH/GSSG is shifted towards plenty of oxidized GSH and subsequently the GSH dependent GST activity is definitely decreased. The decreased GSH concentration as well as the decreased GPX activity reported in our results emphasized the improper GSH/GSSG ratio. Metwally et al.23, engaged the increases in blood GSSG levels during cancer growth to the increased peroxide produced by tumor cells which lead to GSH oxidation and decreased GSH/GSSG ratio. The cells failed to replenish the consumed GSH due to GPX inhibition.
Moreover GST, one of phase II detoxification enzymes, catalyze the conjugation of GSH to a wide variety of endogenous and exogenous electrophilic compounds, protect cells from cytotoxic and carcinogenic agents, remove oxidative stress products and modulate cell proliferation and signaling pathways25. It offers protection against lipid peroxidation by promoting the conjugation of toxic electrophiles with GSH 26. GSH conjugation is the first step in the mercapturic acid pathway that leads to the elimination of toxic compounds. GSTs have evolved with GSH and are abundant throughout most life forms27.Thus inhibition of GST potentiates the cancer transformation and increase the probability of tumorgensis. Srivastava and Mittal28, found that early loss of GSTP1 expression could lead to increased susceptibility to carcinogens, promoting mutation and cancer development.The increase of ROS production may depend on diverse mechanisms, such as the activation of oncogenes, aberrant metabolism, mitochondrial dysfunction, loss of functional p5329. The down regulation of p53 gene (Fig. 2e) could be interpreted in the view of increases in STAT-3 expression (Fig. 2a) causing gene job failure. The cascade inhibitions of tumor suppressor oncogene (p53) smooth the progress of cancer transformation and instigate tumorigenesis. Choi et al.30, pointed to the possible inhibition of p53 gene by the imbalance occurs between FAT10, a target gene of STAT-3 and p53. Further, our results display up regulation of β-catenin expression which could participate in losing of p53 expression. Sadot et al.31, found that, loss of wild type p53 expression in cancers might be driven by excessive accumulation of β-catenin. So, the significant elevation in STAT-3 and β-catenin mRNA in E mice compared to normal mice (Fig. 2d) seemed to be a reflection of collapses in antioxidant/prooxidant equilibrium (Table 2). Worthwhile, the increases in STAT-3 mRNA and MDA in E group it was observed in our previous study32. Evidently, Lipid peroxidation products enhance canonical WNT pathway which act to inactivate destructive complex leading to prevention of the proteosomal degradation of the transcriptional factor β-catenin and promotes its accumulation and nuclear translocation. Once β-catenin translocates into the nucleus, it associates with the T cell factor (TCF) and regulates transcription of the WNT target genes, including VEGF and CTGF. The products of this gene promote angiogensis and suppress apoptosis33. Moreover, the accumulation of lipid peroxides moieties during lipid peroxidation steps are involved in the activation of STAT-334. Constitutively, STAT-3 has been implicated in the induction of resistance to apoptosis. Its role in tumorigenesis is mediated through the expression of various genes that suppress apoptosis, mediate proliferation, invasion and angiogenesis4. Ibrahem et al.5, recorded significant increases in β-catenin mRNA levels follow the stimulation of cells with IL-6 which is the known inducer of STAT-3 expression. Rationally, the frequent observations of collimated increases of STAT-3 and β-catenin in many studies32-35-36assumed the present of certain mutually supporting correlation. Ultimately, the marked increases in tumor volume in EAC bearing mice (Fig. 3) could be connected to the over expressed STAT-3 and β-catenin signal molecules mRNA rather than inhibition of p53 gene. The present of high levels of lipid peroxide products due to heave oxidative status pave to cancer transformation of extra cells and increases cell proliferation and tumor growth. In the present study, we report the identification of a novel inhibitor of STAT-3. Because STAT-3 has been linked with survival, proliferation, chemo-resistance and angiogenesis of tumor cells, its inhibitors have potential for the treatment of cancer. This study investigates the possible inhibitory role of AV on the STAT-3 expression. Our data reveals, administration of AV to EAC bearing mice induced significant decrease in peroxidation products (MDA) whereas the antioxidant parameters undergoes significant increase when compared with EAC mice (Table 2). This could interpreted in the view of antioxidants properties and the capability of AV to lower lipid contents. AV possesses certain antioxidant activity against hydroxyl (OH) and peroxyl radicals. AV and its metabolites could reduce lipoprotein oxidation and ameliorate free radical injuries37-38. The increase of GSH as well as the activities of GPX and GST observed in this study could support the proposition of antioxidant ameliorating effect of AV. Prabha et al.26, revealed that AV administration significantly produce rise in GPX, GST activity and GSH levels. Elevation in the activity of GST, help in subsequent initiation of the apoptotic process in tumor cells, have enormous clinical significance for immunotherapy of various forms of cancer with a completely non toxic therapy23. Further, the reported inhibition in STAT-3 expression in E group after AV administration could be attributed to the blockade of STAT-3 signaling which is supported by the associated up regulation of STAT-3 suppression gene (p53 gene) (Fig. 2). Kim et al.39, showed that, AV treatment up-regulated the levels of the STAT-3-dependent suppressed genes Krüppel-like factor 5 and p53. So, these regulatory effects of AV suppress STAT-3 gene expression and subsequently suppress signaling cascades. Moreover, AV increases the activities of GST could be participate in the observed STAT-3 suppression. Kou et al.25, found that GSTP1, a member of the GST family important in the regulation of the transcriptional activity of STAT-3 and regulator of the cell cycle via epidermal growth factor signaling. Whereas The GSTP1-STAT-3 complex reduces proliferation and arrests the cell cycle by terminating STAT-3 activity. However, the decrease in β-catenin expression might be related to the inhibition of STAT-3 signaling by AV. Ibrahem et al.5, display that, the knockdown of STAT-3 was associated with a fall in β-catenin mRNA levels. Exposure of mice bearing Ehrlich solid tumor to γ-radiation potentiate the action of AV on the sensitive genes, STAT-3 and β-catenin (Fig. 2). In Moustafa et al.32, study, significant decreases in STAT-3 mRNA expression and MDA level were observed in solid tumors bearing mice after exposure to γ-radiation. Radiation therapy is a currently standard treatment for a number of malignancies 40. However, the response of a cancer to radiation is described by its radiosensitivity. Highly radiosensitive cancer cells are rapidly killed by modest doses of radiation41. Further, the radiation effect on the gene expression of STAT-3 might be due to the radiation (3-6 Gy) turn suppressive mechanism (senescence effect) that associated with slowing the production of certain cytokines and growth factor42. Based on the observation in this study, recommended AV which potentiated by radiotherapy for cancer patients to stopping tumour progression.
CONCLUSION
Subsequently, it could postulated that AV possess certain potential in vivo against cancer proliferation which might manifested by inhibition of STAT-3 and β-catenin expression as well as through activation of p53 gene expression. Besides that, radiation exposure could sustain the antitumor effect of AV.
SIGNIFICANCE STATEMENT
This study discovers the inhibitory effect of atorvastatin on the expression of STAT-3 and β-catenin signals molecules that can be beneficial for cancer patients. This study help the researchers to uncover the critical areas of cancer prevention that many researchers were not able to explore. Thus a new theory on blockade of STAT-3 activation and β-catenin expression may be arrived at.
REFERENCES
- Pasterkamp, G. and G.W. van Lammeren, 2010. Pleiotropic effects of statins in atherosclerotic disease. Expert Rev. Cardiovasc. Ther., 8: 1235-1237.
CrossRefDirect Link - Mantha, A.J., J.E.L. Hanson, G. Goss, A.E. Lagarde, I.A., Lorimer and J. Dimitroulakos, 2005. Targeting the mevalonate pathway inhibits the function of the epidermal growth factor receptor. Clin. Cancer Res., 11: 2398-2407.
CrossRefDirect Link - Lee, S.K., Y.C. Kim, S.B. Song and Y.S. Kim, 2010. Stabilization and translocation of p53 to mitochondria is linked to Bax translocation to mitochondria in simvastatin-induced apoptosis. Biochem. Biophys. Res. Commun., 391: 1592-1597.
CrossRefDirect Link - Aggarwal, B.B., A.B. Kunnumakkara, K.B. Harikumar, S.R. Gupta and S.T. Tharakan et al., 2009. Signal transducer and activator of transcription 3, inflammation and cancer: How intimate is the relationship? Ann. N. Y. Acad. Sci., 1171: 59-76.
CrossRefDirect Link - Ibrahem, S., S. Al-Ghamdi, K. Baloch, B. Muhammad and W. Fadhil et al., 2014. STAT3 paradoxically stimulates β-catenin expression but inhibits β-catenin function. Int. J. Exp. Pathol., 95: 392-400.
CrossRefPubMedDirect Link - Yu, H. and R. Jove, 2004. The STATs of cancer-new molecular targets come of age. Nat. Rev. Cancer, 4: 97-105.
CrossRefDirect Link - Saifo, M.S., D.R. Rempinski Junior, Y.M. Rustum and R.G. Azrak, 2010. Targeting the oncogenic protein β-catenin to enhance chemotherapy outcome against solid human cancers. Mol. Cancer, Vol. 9.
CrossRefDirect Link - Ozaslan, M., I.D. Karagoz, I.H. Kilic and M.E. Guldur, 2011. Ehrlich ascites carcinoma. Afr. J. Biotechnol., 10: 2375-2378.
Direct Link - El-Bahy, A.A., K. Abou-Aisha, E. Noaman and L.G. Mahran, 2012. Regression of murine ehrlich ascites carcinoma using tumor-targeting salmonella VNP20009- comparison with the effect of the anticancer drug doxorubicin. Adv. Cancer Res. Treat.
CrossRefDirect Link - Jaganathan, S.K., D. Mondhe, Z.A. Wani, H.C. Pal and M. Mandal, 2010. Effect of honey and eugenol on Ehrlich ascites and solid carcinoma. J. Biomed. Biotechnol., Vol. 2010.
CrossRef - Gupta, M., U.K. Mazumder, R.S. Kumar and T.S. Kumar, 2004. Antitumor activity and antioxident role of Bauhinia racemosa against Ehrlich ascites carcinoma in Swiss albino mice. Acta Pharmacol. Sin., 25: 1070-1076.
Direct Link - Dolai, N., I. Karmakar, R.B. Kumar, A. Bala, U.K. Mazumder and P.K. Haldar, 2012. Antitumor potential of Castanopsis indica (Roxb. ex Lindl.) A. DC. leaf extract against Ehrlich's ascites carcinoma cell. Indian J. Exp. Biol., 50: 359-365.
PubMedDirect Link - Halle, W., 2003. The registry of cytotoxicity: Toxicity testing in cell cultures to predict acute toxicity (LD50) and to reduce testing in animals. Altern. Lab. Anim., 31: 89-198.
PubMedDirect Link - Islam, M., S. Sharma, B. Kumar and T.N. Teknos, 2013. Atorvastatin inhibits RhoC function and limits head and neck cancer metastasis. Oral Oncol., 49: 778-786.
CrossRefPubMedDirect Link - Yoshioka, T., K. Kawada, T. Shimada and M. Mori, 1979. Lipid peroxidation in maternal and cord blood and protective mechanism against activated-oxygen toxicity in the blood. Am. J. Obstet. Gynecol., 135: 372-376.
CrossRefPubMedDirect Link - Ellman, G.L., 1959. Tissue sulfhydryl groups. Arch. Biochem. Biophys., 82: 70-77.
CrossRefPubMedDirect Link - Gross, R.T., R. Bracci, N. Rudolph, E. Schroeder and J.A. Kochen, 1967. Hydrogen peroxide toxicity and detoxification in the erythrocytes of newborn infants. Blood, 29: 481-493.
CrossRefPubMedDirect Link - Habig, W.H., M.J. Pabst and W.B. Jakoby, 1974. Glutathione S-transferases: The first enzymatic step in mercapturic acid formation. J. Biol. Chem., 249: 7130-7139.
CrossRefPubMedDirect Link - Livak, K.J. and T.D. Schmittgen, 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods, 25: 402-408.
CrossRefDirect Link - Mingone, C.J., S.A. Gupte, S. Quan, N.G. Abraham and M.S. Wolin, 2003. Influence of heme and heme oxygenase-1 transfection of pulmonary microvascular endothelium on oxidant generation and cGMP. Exp. Biol. Med. (Maywood), 228: 535-539.
PubMedDirect Link - O'Reilly, M.S., T. Boehm, Y. Shing, N. Fukai and G. Vasios et al., 1997. Endostatin: An endogenous inhibitor of angiogenesis and tumor growth. Cell, 88: 277-285.
CrossRefPubMedDirect Link - Pandya, N.B., P. Tigari, K. Dupadahalli, H. Kamurthy and R.R. Nadendla, 2013. Antitumor and antioxidant status of Terminalia catappa against Ehrlich ascites carcinoma in Swiss albino mice. Indian J. Pharmacol., 45: 464-469.
CrossRefPubMedDirect Link - Metwally, F.M., H.A. El-Mezayen, A.E.A. Moneim and N.E. Sharaf, 2014. Anti-tumor effect of Azadirachta indica (Neem) on murine solid ehrlich carcinoma. Acad. J. Cancer Res., 7: 38-45.
Direct Link - Ali, D.A., N.K.B. El-Din and R.F. Abou-El-Magd, 2015. Antioxidant and hepatoprotective activities of grape seeds and skin against Ehrlich solid tumor induced oxidative stress in mice. Egypt. J. Basic Applied Sci., 2: 98-109.
CrossRefDirect Link - Kou, X., N. Chen, Z. Feng, L. Luo and Z. Yin, 2013. GSTP1 negatively regulates Stat3 activation in epidermal growth factor signaling. Oncol. Lett., 5: 1053-1057.
CrossRefDirect Link - Prabha, S.P., P.N. Ansil, A. Nitha, P.J. Wills and M.S. Latha, 2013. Anti-atherogenic activity of methanolic extract of Gardenia gummifera Linn.F. on high fat diet induced atherosclerosis in rats. Int. J. Pharm. Pharm. Sci., 5: 388-393.
Direct Link - Townsend, D.M. and K.D. Tew, 2003. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene, 22: 7369-7375.
CrossRefDirect Link - Srivastava, D.S.L. and R.D. Mittal, 2005. Free radical injury and antioxidant status in patients with benign prostate hyperplasia and prostate cancer. Indian J. Clin. Biochem., 20: 162-165.
CrossRefDirect Link - Barrera, G., 2012. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol., Vol. 2012.
CrossRefDirect Link - Choi, Y., J.K. Kim and J.Y. Yoo, 2014. NFκB and STAT3 synergistically activate the expression of FAT10, a gene counteracting the tumor suppressor p53. Mol. Oncol., 8: 642-655.
CrossRefPubMedDirect Link - Sadot, E., B. Geiger, M. Oren and A. Ben-Ze'ev, 2001. Down-regulation of β-catenin by activated p53. Mol. Cell. Biol., 21: 6768-6781.
CrossRefPubMedDirect Link - Moustafa, E.M., N.M. Thabet and K.S. Azab, 2016. Boswellic acid disables signal transduction of IL-6-STAT-3 in Ehrlich ascites tumor bearing irradiated mice. Biochem. Cell Biol., 94: 307-313.
CrossRefPubMedDirect Link - Zhou, T., K.K. Zhou, K. Lee, G. Gao, T.J. Lyons, R. Kowluru and J.X. Ma, 2011. The role of lipid peroxidation products and oxidative stress in activation of the canonical wingless-type MMTV integration site (WNT) pathway in a rat model of diabetic retinopathy. Diabetologia, 54: 459-468.
CrossRefPubMedDirect Link - Maziere, C., G. Alimardani, F. Dantin, F. Dubois, M.A. Conte and J.C. Maziere, 1999. Oxidized LDL activates STAT1 and STAT3 transcription factors: Possible involvement of reactive oxygen species. FEBS Lett., 448: 49-52.
CrossRefPubMedDirect Link - Anand, M., R. Lai and P. Gelebart, 2011. β-catenin is constitutively active and increases STAT3 expression/activation in anaplastic lymphoma kinase-positive anaplastic large cell lymphoma. Haematologica, 96: 253-261.
CrossRefPubMedDirect Link - Fragoso, M.A., A.K. Patel, R.E.I. Nakamura, H. Yi, K. Surapaneni, A.S. Hackam, 2012. The Wnt/β-catenin pathway cross-talks with STAT3 signaling to regulate survival of retinal pigment epithelium cells. PLOS ONE, Vol. 7, No. 10.
CrossRefDirect Link - Mehany, H.A., A.M. Abo-Youssef, L.A. Ahmed, E.S.A. Arafa and H.A. Abd El-Latif, 2013. Protective effect of vitamin E and atorvastatin against potassium dichromate-induced nephrotoxicity in rats. Beni-Suef Univ. J. Basic Applied Sci., 2: 96-102.
CrossRefDirect Link - Khodayar, M.J., M. Kiani, A.A. Hemmati, A. Rezaie-Majd and M.R. Zerafatfard et al., 2014. The preventive effect of atorvastatin on paraquat-induced pulmonary fibrosis in the rats. Adv. Pharm. Bull., 4: 345-349.
CrossRefPubMedDirect Link - Kim, B.H., S. Han, H. Lee, C.H. Park and Y.M. Chung et al., 2015. Metformin enhances the anti-adipogenic effects of atorvastatin via modulation of STAT3 and TGF-β/Smad3 signaling. Biochem. Biophys. Res. Commun., 456: 173-178.
CrossRefPubMedDirect Link - Becker, K.P. and J. Yu, 2012. Status quo-standard-of-care medical and radiation therapy for glioblastoma. Cancer J., 18: 12-19.
CrossRefPubMedDirect Link - Yu, Y.C., P.M. Yang, Q.Y. Chuah, Y.H. Huang, C.W. Peng, Y.J. Lee and S.J. Chiu, 2013. Radiation-induced senescence in securin-deficient cancer cells promotes cell invasion involving the IL-6/STAT3 and PDGF-BB/PDGFR pathways. Sci. Rep., Vol. 3.
CrossRefDirect Link