
I.T. Silva
Department of Pharmaceutical Sciences, Universidade Federal de Santa Catarina, Florianopolis, SC, Brazil
Marina R. Teixeira
Department of Pharmaceutical Sciences, Universidade Federal de Santa Catarina, Florianopolis, SC, Brazil
Karen L. Lang
Department of Chemistry, Universidade Federal de Santa Catarina, Florianopolis, SC, Brazil
Tatiana da R. Guimaraes
Department of Pharmaceutical Sciences, Universidade Federal de Santa Catarina, Florianopolis, SC, Brazil
Sabine E. Dudek
Institute of Molecular Virology (IMV), Center of Molecular Biology of Inflammation (ZMBE), University of Muenster, D-48149 Muenster, Germany
Fernando J. Duran
UMYMFOR-Department of Organic Chemistry, Universidad de Buenos Aires, Buenos Aires, Argentina
Stephan Ludwig
Institute of Molecular Virology (IMV), Center of Molecular Biology of Inflammation (ZMBE), University of Muenster, D-48149 Muenster, Germany
Miguel S.B. Caro
Department of Chemistry, Universidade Federal de Santa Catarina, Florianopolis, SC, Brazil
Eloir P. Schenkel
Department of Pharmaceutical Sciences, Universidade Federal de Santa Catarina, Florianopolis, SC, Brazil
Claudia M.O. Simoes
Department of Pharmaceutical Sciences, Universidade Federal de Santa Catarina, Florianopolis, SC, Brazil
International Journal of Cancer Research
Year: 2013 | Volume: 9 | Issue: 2 | Page No.: 54-68







ABSTRACT
Cancer represents a major public health problem from all over the world and the lung carcinoma is the leading cause of cancer death. In this sense, the aim of the present study was to examine the cytotoxic effects of a novel Cucurbitacin (Cuc1) isolated from Wilbrandia ebracteata on human non-small-cell lung cancer (A549). In order to achieve this aim, the cell proliferation was measured by MTT assay and actin cytoskeleton was stained by rhodamine-phalloidin, whereas, the cell cycle distribution and apoptosis induction were quantified using flow cytometry. The signal transduction profiling of Cuc1 treated cells, as well as the levels of apoptotic proteins were analyzed by Western blotting. Cuc1 significantly inhibited cell growth showing IC50 values of 13.5±1.8 and 3.8±0.4 μM for 48 and 72 h of treatment, respectively. Additionally, Cuc1 arrested cell cycle at G2/M phase, disrupted the actin dynamics and induced apoptosis since the amount of apoptotic cells increased from 5.18±0.585% in the untreated cells to 73.82±0.545% in the treated cells. Detailed analysis on the mechanism of action revealed that Cuc1 inhibited the phosphorylation of Protein Kinase B (PKB/AKT) and Signal Transducer and Activator of Transcription (STAT3) signaling pathways, down-regulating the expression of Bcl-2 and consequently inducing cytochrome c release from the mitochondria to the cytosol. These results suggest that the Cuc1 could be a potential candidate for cancer chemotherapy agent.
PDF Abstract XML References Citation
Received: March 12, 2013;
Accepted: April 18, 2013;
Published: October 28, 2013
How to cite this article
I.T. Silva, Marina R. Teixeira, Karen L. Lang, Tatiana da R. Guimaraes, Sabine E. Dudek, Fernando J. Duran, Stephan Ludwig, Miguel S.B. Caro, Eloir P. Schenkel and Claudia M.O. Simoes, 2013. Proliferative Inhibition and Apoptotic Mechanism on Human Non-small-cell
Lung Cancer (A549 Cells) of a Novel Cucurbitacin from Wilbrandia ebracteata
Cogn. International Journal of Cancer Research, 9: 54-68.
DOI: 10.3923/ijcr.2013.54.68
URL: https://scialert.net/abstract/?doi=ijcr.2013.54.68
DOI: 10.3923/ijcr.2013.54.68
URL: https://scialert.net/abstract/?doi=ijcr.2013.54.68
INTRODUCTION
Cancer can be considered a major public health problem all around the world. In the United States, 25% of all deaths are related to cancer. Only in 2011, were estimated 221,130 new cases and 156,940 deaths related to lung cancer specifically. Worldwide, the non-small-cell lung carcinoma (NSCLC) is the leading cause of cancer death in both men and women (Han and Roman, 2010; Siegel et al., 2011). In spite of quick advances in diagnostic and surgical techniques, lung cancer remains one of the hardest treatable human cancers. Until today treatment modalities are ineffective and new therapies are necessary to reduce the effects of the increasing incidence in this type of cancer (Kim et al., 2003).
Apoptosis is a form of programmed cell death that permits to eliminate unwanted, redundant, or mutated cells from organisms, without causing damage to the cellular microenvironment (Russo et al., 2006). In cancer cells, multiple genetic mutations and the overall cellular stresses of malignant transformation are associated with substantial pro-apoptotic activity (Call et al., 2008). However, cancer can persist and dysregulation of apoptosis might be essential for the survival of many cancers providing inherent resistance to chemotherapeutic agents (Tan and White, 2008; Sayers, 2011). Thus, a rational approach for treating cancer is the modulation of apoptosis pathway by targeting components of the apoptotic mechanism and its regulators in order to reestablish the apoptotic function (Nicholson, 2000; Call et al., 2008; Indran et al., 2011).
Plants synthesize a wide variety of biologically active compounds and for that reason there is a growing interest in their use as a source of new anticancer drugs (Lee et al., 2011). Cucurbitacins, for instance, are a group of diverse tetracyclic triterpenoid molecules, predominantly found in different species of the Cucurbitaceae family that are highly bitter and toxic. Their structural configuration compromises tetracyclic cucurbitane nucleus skeleton, with a variety of oxygenation functionalities at different positions (Chen et al., 2005). Plants containing cucurbitacins have been used for centuries with ethnomedical purposes (Lee et al., 2010) and several compounds of this group have shown antitumor, anti-inflammatory and hepatoprotective effects (Jayaprakasam et al., 2003; Rios et al., 2012). They have also shown strong activity against tumor expansion (Sun et al., 2010; Yasuda et al., 2010) as demonstrated for cucurbitacins B, D, E and I that inhibited the growth of several cancer cell lines in in vitro and in vivo models (Su et al., 2008; Wakimoto et al., 2008; Yin et al., 2008; Chen et al., 2010; Dong et al., 2010; Sun et al., 2010; Yasuda et al., 2010; Zhang et al., 2010; Hsu et al., 2011; Ishdorj et al., 2011).
Recently, novel cucurbitacins obtained from Wilbrandia ebracteata Cogn. (Cucurbitaceae) have been described (Lang et al., 2011) as well as some semi-synthetic derivatives (Lang et al., 2012) that exhibited promising cytotoxic activity against different human cancer cell lines. The present study examined the impact of one of these new cucurbitacins (2β, 16α, 2R-trihydroxy-10α, 17α-cucurbit-5, 25-dien-3, 11, 22-trione, Cuc 1) (Fig. 1) on non-small-cell lung cancer (A549 cells), including the evaluation of its effects on cell growth, cell cycle distribution, apoptosis, morphological changes and expression of regulatory proteins involved in such processes.
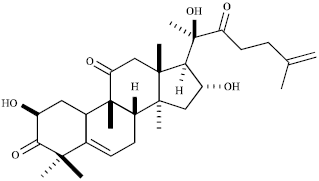 | |
| Fig. 1: | Structure of a novel cucurbitacin (Cuc 1) isolated from Wilbrandia ebracteata roots |
MATERIALS AND METHODS
Isolation and identification of Cuc 1: Roots of Wilbrandia ebracteata Cogn. were purchased from Lohmann Company Ltd. and authenticated by Prof. Dr. Sergio A.L. Bordignon (Unilassale, Canoas, RS, Brazil). Dried and powderedroots (1.5 kg) were extracted with dichloromethane (DCM) at room temperature for 72 h. The DCM extract (9 g) was subjected to vacuum liquid column chromatography on silica gel using hexane with increasing amounts of ethyl acetate (20-100%) to afford eight fractions (F1 to F8). The fractions F3 and F4 (500 mg) were combined and subjected to column chromatography using silica gel as adsorbent and hexane/ethyl acetate 40% as mobile phase providing Cuc 1 (10 mg) which was identified by spectroscopic methods as described previously (Lang et al., 2011).
Cell line: The human non-small-cell lung cancer (A549 cells) was kindly provided by Dr. Rosina Gironès from Microbiology Department of University of Barcelona, Spain. A549 cells (ATCC-CCL185) were grown in Minimal Essential Medium supplemented with 10% fetal bovine serum, 100 U mL-1 penicillin G and 100 μg mL-1 streptomycin in a humidified 5% CO2 atmosphere at 37°C.
Cell viability assay: The effect of Cuc 1 treatment on the viability of A549 cells was determined by MTT assay [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] (Sigma, MO, USA) based on the ability of live cells to cleave the tetrazolium ring to a molecule that absorb at 540 nm (Mosmann, 1983). Briefly, cells were plated in 96-well culture plates (1¯104 cells/well). After 24 h, seeding cells were treated with different concentrations of Cuc 1. After 48 and 72 h of treatment at 37°C, the medium was replaced by 50 μL of MTT reagent (1 mg mL-1) and cells were further incubated at 37°C for 4 h. The MTT solution was removed, 100 μL of dimethyl sulfoxide was added to each well, followed by reading on a scanning multiwell spectrophotometer (Infinite 1200 TECAN, Grödje, Austria). The 50% inhibition concentration (IC50) was defined as the concentration that inhibited cell proliferation by 50% when compared to untreated controls (=untreated cells). Paclitaxel (Sigma, MO, USA) (at 0 to 10 μM) was used as positive control. Final solvent concentration showed no interference with cell growth.
Cell cycle analysis by flow cytometry: A549 cells (5x105/six-well) were treated with Cuc 1 for 24 h and harvested by 0.25% trypsin. Then cells were washed twice with Phosphate-buffered Saline (PBS), centrifuged at 500x g for 5 min, fixed in 70% ethanol at -4°C. After fixation, the cells were treated with 50 μg mL-1 RNase and stained with 100 μg mL-1 Propidium Iodide (PI) in the dark. Flow cytometry analyses were carried out on a FACS CAnto II instrument (Becton Dickinson, USA). The population of cells in each cell-cycle phase was determined using FlowJo 8.6.3 software (Tree Star, Inc., Ashland, USA).
Apoptosis analysis by flow cytometry: Phosphatidylserine (PS) exposed on the outside of apoptotic cells was detected by Annexin V-FITC and PI double-staining by using a detection kit purchased from Sigma (MO, USA). Briefly, adherent A549 cells (5x105/six-well) were treated with Cuc 1 for 12 h. Cells were harvested and rinsed twice with PBS (pH 7.4) followed by Annexin V-FITC and PI labeling according to the manufacturer’s instructions. The stained apoptotic cells were analyzed by flow cytometer (FACS CAnto II, Becton Dickinson, USA).
Cytoskeletal and nuclei staining: F-actin detection was performed on A549 cells grown on coverslips placed into a six-well plate. Cells were treated with Cuc 1 for 12 h and fixed by 4% (w/v) paraformaldehyde in PBS for 15 min at room temperature, washed in PBS and permeabilized with 0.5% (v/v) Triton X-100 for 30 min at RT. After three-times washing, cells were incubated with TRITC-labeled-phalloidin (Invitrogen, Carlsbad, USA) for 40 min in the dark. Preparations were washed and stained with Hoechst (Invitrogen, Carlsbad, USA) for 5 min to detect nuclei, washed and finally mounted in mounting medium (80% glycerol in PBS). Confocal images were collected on a Leica DMI6000 B microscope (Leica Microsystems, Wetzlar, Germany).
Caspase assay: Caspases -3, -8 and -9 protease activity was determined by using commercially available kits (Millipore, MA, USA) according to the manufacturer’s instructions. The tests are based on the spectrophotometric detection of the chromophore p-nitroanaline (pNA) after cleavage from the caspase substrate (a caspase-specific peptide conjugated to pNA). Briefly, adherent A549 cells (5x105/six-well) were treated with Cuc 1 for 12 h and harvested by 0.25% trypsin. The cells were resuspended in 50 μL of chilled cell lysis buffer, centrifuged for 1 min (10,000xg) and then the supernatants were transferred (cytosolic extract) to a fresh tube and put on ice for immediate assay. The cell lysates were tested for protease activity by the addition of a labelled caspase substrate (DEVD-pNA for caspase-3 activity, IETD-pNA for caspase-8 activity and LEHD-pNA for caspase-9) and incubated at 37°C for 2 h. pNA absorbance was quantified using a spectrophotometer (Infinite 1200 TECAN, Grödje, Austria) at a wavelength of 405 nm. Fold-increase in caspase-activity can be determined by comparing the OD reading from the induced apoptotic sample with the level of the uninduced control.
Western blotting analysis: Adherent A549 cells (1x106/six-well) were treated with Cuc 1 for 12, 24 or 48 h. To evaluate the influence of Cuc 1 on Akt-, MAPKs- and NFκB- signaling pathways, TNFα stimulation (30 ng mL-1, 20 min) was used as a pathway inducer. Whole cell lysates were harvested after lysis with RIPA buffer containing proteases and phosphatases inhibitors and then centrifuged at 10,000x g for 10 min at 4°C. The supernatants were collected and the protein concentrations were determined using the Bradford assay. Equivalent amounts of protein were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by electrotransfer to a nitrocelullose membrane (Schleicher and Schuell, Dassel, Germany). After blocking, membranes were incubated overnight with appropriate primary antibodies. After incubation with the corresponding secondary antibodies conjugated to horseradish peroxidase, protein bands were revealed by using Pierce ECL substrate (Thermo Scientific, MA, USA), according to the manufacturer’s protocol.
Cytosolic fraction was prepared for detecting cytochrome c release. For this, treated and untreated A549 cells were washed twice with cold PBS and lysed with 200 μL of extraction buffer, containing proteases inhibitors, for 10 min on ice. Next, cells were homogenized by 10 passages through a 26-gauge needle. Homogenates were centrifuged at 1,000x g for 5 min to remove unbroken cells and nuclei. The supernatant fraction was centrifuged at 12,000x g for 30 min at 4°C. The resulting supernatant containing the cytosolic fraction was collected and the protein concentrations were determined using the Bradford assay. Then equivalent amounts of protein were analyzed by Western/ECL analyses as described above. Protein bands were quantified using the Advanced Image Data Analyzer Software (AIDA, Raytest, GmbH, Straubenhardt, Germany). The total band densities were measured against the local background. Results were presented as normalized fold changes in relation to control.
The antibodies against cleaved caspase-3, Akt, phospho-p38 MAPK (Thr180/Tyr182) (3D7), phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204), phospho-NFκB p65 (Ser536)(93H1) and NFκB p65 were all purchased from Cell Signaling Technology, MA, USA. The phospho-STAT3 and Bcl-2 antibodies were obtained from Millipore, MA, USA whereas anti-cytochrome c and anti-JNK/SAPK (pT183/pY185) were purchased from Becton Dickinson, NJ, USA. The anti-p38α, IκBα and JNK 1/3 were obtained from Santa Cruz Biotechnology, CA, USA and the phospho-Akt/PKB [pS473] antibody was acquired from Invitrogen, CA, USA. Antibodies against β-actin (Millipore, MA, USA) and α-tubulin (Sigma-Aldrich, MO, USA) served as controls for equal loading.
Statistical analysis: The results were expressed as the Mean±SD of three independent experiments, unless otherwise stated. Statistical analyses were performed by one-way ANOVA followed by Tukey’s post hoc test (p<0.05). Calculations were performed with GraphPad Prism software 5.1 version (GraphPad, USA).
RESULTS
Cuc 1 inhibits A549 cells growth in vitro: To evaluate the cytotoxicity of Cuc 1 on human non-small lung cancer cells, A549 cells were initially treated with different concentrations of Cuc 1 at different time points and cellular proliferation was evaluated using the MTT assay. Treatment with this compound inhibited cellular proliferation in a concentration and time-dependent manner (Fig. 2). Their IC50 values were 13.5±1.8 and 3.8±0.4 μM for 48 and 72 h, respectively.
Cuc 1 induces cell cycle arrest at G2/M phase: Most anticancer agents exhibit their effects on tumor cell growth by inducing cell cycle arrest and apoptosis. To gain insights into the mechanism by which cell proliferation is reduced, the effects of Cuc 1 on distribution of cell cycle phases in a cell population was investigated by Fluorescence-activated Cell Sorting (FACS) analysis (Fig. 3a). The untreated control cells showed a typical distribution in G0/G1, G2/M and S phase, but a 24 h exposure of A549 cells to 15 and 30 μM of Cuc 1 caused a significant enrichment of cells in G2/M phase in a concentration-dependent manner. An increase from 17.06-32.42 and 37.72% cells in G2/M phase was detected after the treatment with 15 and 30 μM of Cuc 1, respectively (p<0.0001 vs. control).
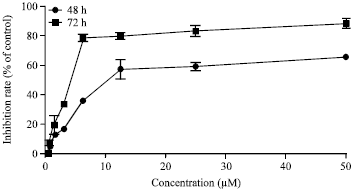 | |
| Fig. 2: | Cell growth inhibition by Cuc 1. Human non-small cell lung cancer cells (A549 cells) were treated with different concentrations of the Cuc 1 for 48 and 72 h. The growth inhibition effects were determined by MTT assay and the IC50 was calculated by Graph Pad Prism 5.1. Values were averaged expressed as a percentage relative to the untreated controls. Values indicate the Mean±SD in triplicate tests |
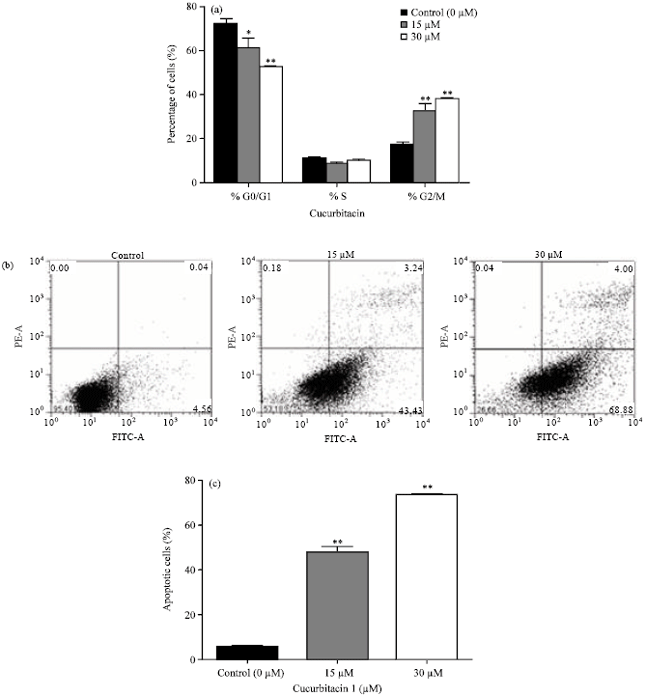 | |
| Fig. 3(a-c): | Effects of Cuc 1 on cell cycle distribution and apoptosis of A549 cells. (a) A549 cells (5x105) were treated with 15 and 30 μM and analyzed after 24 h by DNA flow cytometry. The values indicate the percentage of A549 cells in the indicated phases of the cell cycle (G0/G1, S and G2/M). *p<0.01 and **p<0.0001 as compared with control, (b) The cells were treated with 15 and 30 μM for 12 h, stained with Annexin V/PI and submitted to flow cytometry for the analysis of apoptotic cells proportion. The horizontal (FITC-A) and the vertical (PE-A) axes represent labeling with Annexin V and PI, respectively, (c) Graphic representation of data obtained from Annexin V and PI staining assay. **p<0.0001 as compared with control. The values represented means of three independent experiments and SD. The most representative results of three independent experiments are shown here |
This was followed by a reduction in G0/G1 phase cells (72.14% cells in the controls, decreasing to 61.77 and 52.48% cells after the treatment with 15 and 30 μM of Cuc 1, respectively). These results indicated that the compound induces in vitro growth inhibition of A549 cells by causing G2/M cell cycle arrest.
Cuc 1 induces apoptosis: As the treatment of A549 cells with Cuc 1 inhibited the cell growth it was further analyzed whether this effect might be related to apoptosis induction. One of the earliest events of apoptosis is the loss of plasma membrane polarity accompanied by translocation of Phosphatidylserine (PS) from the inner to outer membrane leaflets, thereby exposing PS to the external environment. The phospholipids-binding protein Annexin V has a high affinity to PS thereby labeling cells with externally exposed PS which correlates with loss of membrane polarity during apoptosis, but precedes the complete loss of membrane integrity that accompanies later stages of cell death resulting either in apoptosis or necrosis (Van Genderen et al., 2006). In contrast, PI can only enter into cells after complete loss of membrane integrity. Thus, dual staining with Annexin V and PI allows clear discrimination between healthy cells (low FITC fluorescence and low PI fluorescence, LL), early apoptotic cells (high FITC fluorescence and low PI fluorescence, LR), late apoptotic cells (high FITC fluorescence and high PI fluorescence, UR) and necrotic cells (low FITC fluorescence and high PI fluorescence, UL). A 12 h exposure of A549 cells to 15 and 30 μM of Cuc 1 increased the percentage of apoptotic cells from 5.18% (LR+UR) in the controls to 47.96 and 73.82% in treated cells, respectively (p<0.0001 vs. control) (Fig. 3b and c). Thus, treatment of A549 cells with Cuc 1 induces apoptosis.
Cuc 1 alters cell morphology and actin organization: Under light microscopy, A549 cells showed drastic alterations in their overall morphology after exposure to Cuc 1 including reduced cytoplasmatic volume and cell rounding (data not shown). Some of these morphological changes could be explained by the disruption of actin cytoskeleton homeostasis by cucurbitacins, as it has been previously described (Tannin-Spitz et al., 2007; Wakimoto et al., 2008; Lee et al., 2010). Therefore, the effects of Cuc 1 treatment on the cytoskeletal network were assessed. For this purpose, untreated and treated A549 cells were stained with TRITC-labeled-phalloidin which binds selectively to F-actin. As shown in Fig. 4a, the untreated cells did not show a prominent polymerized, filamentous actin. However, 12 h exposure to 15 and 30 μM of Cuc 1 induced accumulation of F-actin into globular aggregates in the cytoplasm near to the nuclei as it was demonstrated by the intense fluorescent accumulation of rhodamine-phalloidin staining (Fig. 4b and c). In addition, when cell nuclei were visualized with Hoechst staining, further morphological alterations in Cuc 1 treated A549 cells were observed such as chromatin condensation, nuclear fragmentation and appearance of multinucleated cells. These observations further underline the potential of Cuc 1 to induce cell death by apoptosis.
Cuc 1 induces apoptosis via activated caspases-dependent pathway: Caspases are well known executors of the apoptotic process through their ability to cleave several cellular substrates. To evaluate the effects of Cuc 1 on the activation of the caspase-3, -8 and -9 in A549 cells, the cytosolic extracts of treated or untreated control cells were incubated with different caspase-specific substrates. As shown in Fig. 5, Cuc 1 caused a significant increase in the proteolytic activity of caspases -3 and -9 after 12 h of treatment with the higher concentration (30 μM) compared to untreated control cells. Thus the morphological apoptosis induction observed correlates well with caspase activation.
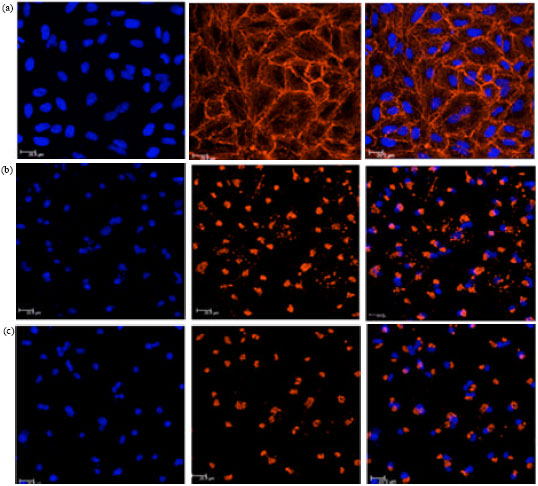 | |
| Fig. 4(a-c): | Effects of Cuc 1 on F-actin organization and nuclear fragmentation by confocal microscopic analysis. A549 cells were either untreated (a) or treated with 15 μM (b) and 30 μM (c) of Cuc 1 for 12 h, fixed and stained with Hoechst (left panels) and TRITC-labeled-phalloidin (middle panels). Overlay images are shown on the right side |
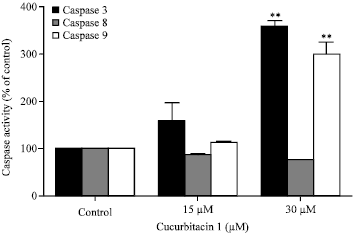 | |
| Fig. 5: | Activation of various caspases in A549 cells treated with Cuc 1. A549 cells were incubated with 15 and 30 μM for 12 h. Cytosolic fraction of cells was analyzed for changes in the activity of caspase-3, caspase-8 and caspase-9. The date represent Means±SD of three independent experiments. **p<0.0001 as compared with control |
Cuc 1 suppresses STAT3 activation: Persistent STAT3 (signal transducer and activator of transcription 3) activation in cancer cells seems to confer their normal physiological function in controlling cell growth, survival, angiogenesis and immune responses. Conversely, the blockage of STAT3 signaling pathway in cancer cells has been shown to induce apoptosis, inhibit cell proliferation, suppress angiogenesis and stimulate immune responses and for these reasons the STAT proteins are emerging as ideal targets for cancer therapy (Yu and Jove, 2004). Previous studies have shown that cucurbitacins suppress tumor growth and induce apoptosis in A549 cells by inhibiting STAT3 signaling (Sun et al., 2005). To determine the relationship between the cytotoxic activity of Cuc 1 and STAT3 phosphorylation, the p-STAT3 expression in A549 cells treated with Cuc 1 was investigated by Western blotting analysis. It was shown that 30 μM of Cuc 1 reduced the STAT3 phosphorylation almost completely after 12 h of treatment compared to the untreated control cells (Fig. 6a).
Cuc 1 alters the expression of apoptosis-related proteins: In order to confirm the ability of Cuc 1 to activate caspase-3, as previously demonstrated by the cleavage of a caspase-3-specific substrate, the accumulation of the active cleaved form in A549 cells was investigated by Western blotting analysis. As shown in Fig. 6b, the treatment of A549 cells with both concentrations of Cuc 1 for 12 h increased the amount of active caspase-3 compared to untreated control cells. The active caspase-3 proteolytically cleaves and activates other caspases, as well as relevant targets in the apoptotic cells. Taken together, these results confirm the involvement of this enzyme in the mechanism of cell death mediated by Cuc 1.
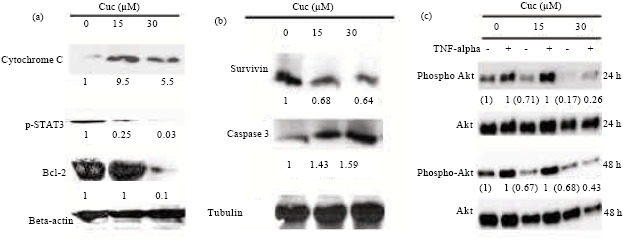 | |
| Fig. 6: | Expression of apoptosis-related proteins and down-regulation of the Akt signaling pathway in unstimulated (TNFα-) and stimulated (TNFα+) A549 cells. (a and b) Western blotting analysis of cytochrome C, p-STAT3, Bcl-2, caspase 3 (cleaved form) and survivin in A549 cells untreated (lane 1) or treated with 15 μM (lane 2) and 30 μM (lane 3) of Cuc 1 for 12 h. β-actin or α-tubulin was used as the loading control. (c) Western blotting analysis of phospho-Akt in A549 cells untreated (lanes 1 and 2) or treated with 15 μM (lanes 3 and 4) and 30 μM (lanes 5 and 6) of Cuc 1 for 24 and 48 h. Total amount of Akt was used as the loading control. Data were representative of three independent experiments |
Cytochrome c release is one of the limiting factors in caspase-9 activation and represents a coordinating control step in the apoptotic process. Since Cuc 1 induced caspase-9 activity (Fig. 5), its ability to trigger cytochrome c release in A549 cells was analyzed. Indeed, after 12 h of treatment with Cuc 1, release of mitochondrial cytochrome c into the cytosol of A549 cells was detected (Fig. 6a). In addition, the treatment with 30 μM of Cuc 1 reduced the expression of the anti-apoptotic protein Bcl-2 in A549 cells (Fig. 6a).
Akt, also known as Protein Kinase B (PKB), is a key player in regulating cellular growth and survival. In the past decade the role of Akt in cancer has increased enormously and it is now evident that activation of Akt is one of the most common molecular alterations in human malignancy. Therefore, Akt has become an increasingly important target of drug development efforts and several inhibitors are now entering clinical trials (Altomare and Testa, 2005; Radisavljevic, 2008). It has been known that disruption of actin cytoskeleton network reduces Akt signaling, leading to a reduced expression of anti-apoptotic proteins such as survivin which ends up in both G2/M arrest and apoptosis (Mosmann, 1983; Liang et al., 2003). Since cell transition to a round shape morphology and disorganization of actin filamentous was observed after Cuc 1 treatment (Fig. 4), the activation state or expression of Akt and survivin was analyzed. Indeed, Cuc 1 treatment for 24 and 48 h caused a significant reduction in Akt activation detected by its phosphorylation on Ser273 in both pre-activated (stimulated) and unstimulated cells, whereas the total amount of Akt remained unaffected (Fig. 6b). In addition, a 24 h treatment with Cuc 1 also led to a reduced survivin expression (Fig. 6c). However, when analyzing the activation state of proteins of other signaling pathways such as ERK, p38, JNK, p65 as well as the degradation of IκBα, no significant changes were observed after treatment with Cuc 1 (data not shown). Taken together, these data demonstrate that Cuc 1 induces apoptosis in A549 cells via a reduced activation of Akt signaling pathway.
DISCUSSION
Natural products provide a rich source of chemopreventive and chemotherapeutic agents (Shukla and Singh, 2011). Cucurbitacins refer to a group of tetracyclic triterpenoids initially identified in the plant family of Cucurbitaceae. In traditional medicine cucurbitacin-containing plants have been used against skin affections, as purgative and emetic and to treat inflammatory conditions (Lewis and Elvin-Lewis, 1977).
This study evaluated a new cytotoxic cucurbitacin named Cuc 1 (Fig. 1), isolated for the first time by Lang et al. (2011) from Wilbrandia ebracteata Cogn. roots. Therefore, the antiproliferative effects of Cuc 1 on human non-small-cell lung cancer cells (A549 cells) as well as the molecular mechanism of its cytotoxic properties were investigated.
MTT analysis showed significant inhibition of A549 cells viability in vitro in a concentration and time-dependent manner (Fig. 2). Cuc 1 treatment inhibited cell growth showing IC50 values of 13.5±1.8 and 3.8±0.4 μM for 48 and 72 h, respectively. In addition, this compound showed higher selectivity towards A549 cells, when compared to healthy cells (human gingival fibroblasts) presenting an IC50 value of 132.95±24.11 μM after 48 h exposure to these cells (data not shown). It is well known that most anticancer agents exhibit their inhibitory effects on tumor cell growth by inducing cell cycle arrest and apoptosis. In order to better characterize the mechanism underlying the observed inhibitory activity of Cuc 1 on A549 cells, a set of experiments on cell cycle distribution and apoptosis detection by flow cytometry was performed. Here A549 cells treated with Cuc 1 were arrested at the G2/M phase of cell cycle with decreased cell population in G0/G1 (Fig. 3a), suggesting that this new compound inhibits cell growth via blocking the division cell cycle at the G2/M phase. The results of the present study are in agreement with recent reports which have demonstrated that this group of natural compounds induces G2/M arrest and apoptosis in other human cancer cell lines in vitro (Tannin-Spitz et al., 2007; Li et al., 2010; Yasuda et al., 2010).
Phosphatidylserine externalization is a hallmark of early steps in apoptosis. As Cuc 1 was shown to induce cell death it was further analyzed whether this special event is induced upon Cuc 1 treatment. By flow cytometry analysis with Annexin V/PI staining was confirmed that it induced apoptosis in a concentration-dependent manner (Fig. 3b and 3c).
Confocal microscopy analyses with rhodamine-phalloidin and Hoechst fluorescent staining revealed that Cuc 1 exposure led to the development of morphologically altered and multinucleated A549 cells at both tested concentrations (Fig. 4). These morphological changes are typical for apoptosis. One of the most obvious morphological effects of Cuc 1 on A549 cells was the disruption of F-actin cytoskeleton as showed by the altered appearance of rhodamine-phalloidin stainable material. This is in complete consistence with the current knowledge of cucurbitacins. Some scholars have reported that cucurbitacins can directly modulate the actin cytoskeleton. Duncan et al. (1996) demonstrated that cucurbitacin E acts as a potent disruptor of cytoskeletal integrity by increasing the filamentous or polymerized actin fraction in prostate carcinoma cells. Other studies carried out with cucurbitacin B also showed the aggregation of F-actin in various human cancer cell lines (Haritunians et al., 2008; Wakimoto et al., 2008; Yin et al., 2008).
Multinucleation is a consistently reported morphological alteration in cancer cell cultures that are treated with cucurbitacins (Siqueira et al., 2009; Lee et al., 2010). The final step in cell division, in which the cytoplasm is divided to form two daughter cells, is known as cytokinesis. Since cytokinesis involves the assembly and disassembly of actin filaments, compounds that interfere with actin polymerization or its spatial organization will block this process and generate multinucleated cells by uncoupling nuclear and cytoplasmic division. Therefore, molecules acting on the actin cytoskeleton of tumor cells and thus inhibiting cell division and cell proliferation, may be of high therapeutic value. Currently, growing evidence indicates that cytoskeletal components are involved in the apoptotic cascade thereby regulating cell survival (Cabado et al., 2004). In a previous study, it was found that cytochalasin B which causes cytoskeletal disruption, also inhibited cell proliferation, leading to arrest in G2/M phase and finally apoptosis induction (Liang et al., 2003). These effects are similar to those observed in this study for Cuc 1. Thus, one can propose that the observed effects are mediated, at least partially, by a modification of the cytoskeleton network causing changes in cell morphology, leading to reduction of Akt phosphorylation, G2/M arrest and apoptosis induction.
In the regulation of apoptosis, several caspases play important roles (Hengartner, 2000). They are organized into initiator or effector caspases, due to the role they play in apoptosis induction. It is important to state that activation of caspases is a hallmark of promoting apoptosis in response to death inducing signals originated mainly from cell surface receptors or mitochondria. It has also been shown that caspase-3 activation, the major effector caspase, requires the activation of initiator caspases such as caspase-8 or -9 in response to the different pro-apoptotic signals (Budihardjo et al., 1999). Herein, the activation of the effector caspases-3 was observed in response to the initiator caspase-9 in A549 cells treated with Cuc 1. This suggests that the intrinsic pathway might be involved in apoptosis induced by this novel compound.
Signal transducer and activator of transcription protein 3 (STAT3) is a latent cytosolic transcription factor that transfers signals from the cell membrane directly to the nucleus. STAT3 is constitutively activated in multiple human cancers including ovarian, breast, prostate and lung cancer. It plays a key transcriptional role in cancer cell progression, differentiation and survival by up-regulating several genes, including those that encode for anti-apoptotic proteins and some cell cycle regulators (Yu and Jove, 2004; Fletcher et al., 2009). Thus, since most of the chemotherapeutic strategies aim to initiate apoptosis, it is now generally accepted that STAT3 represents a valid target for novel anticancer drug design (Al Zaid Siddiquee and Turkson, 2008). Although initial interest in cucurbitacins as potential anticancer drugs declined for decades, recent discoveries showing that they are strong STAT3 inhibitors have regained the attention of the pharmaceutical industry one more time. Several studies have suggested that cucurbitacins exert inhibitory effects against many human cancer cell lines via suppression of STAT3 phosphorylation (Sun et al., 2005; Thoennissen et al., 2009; Chan et al., 2010; Liu et al., 2010; Sun et al., 2010). In this study, the evaluation of the phosphorylation state of STAT3 in A549 cells by using Western blotting analysis demonstrated the anti-STAT3 activity of Cuc 1 (15 and 30 μM), as shown in Fig. 6a. These findings suggest that the inhibition of STAT3 phosphorylation may be associated to the induction of apoptosis by Cuc 1.
To determine the expression changes of STAT3 target genes involved in cell apoptosis, the expression of Bcl-2 was analyzed showing that Cuc 1 treatment decreased Bcl-2 expression in a concentration-dependent manner (Fig. 6a).
The Bcl-2 family members also play a critical role in the regulation of apoptosis, comprising both pro-apoptotic molecules (Bax, Bcl-Xs, Bak, Bid, Bad, Bim, Bik) and anti-apoptotic molecules (Bcl-2, Bcl-XL, Bcl-W, Mcl-1, A1). These molecules control the release of mitochondrial cytochrome c by modulating the permeability of the outer mitochondrial membrane (Lessene et al., 2008). Herein, Cuc 1 treatment induced cytochrome c release from mitochondria to the cytosol (Fig. 6a) and it is well known that cytochrome c release is necessary for the activation of caspase-9 that initiates the caspases cascade. These results also suggest that the mitochondrial pathway plays an important role in the apoptosis of A549 cells induced by Cuc 1. In addition, activated Akt is a well-established survival factor exerting anti-apoptotic activity, in part by preventing the release of cytochrome c from the mitochondria and inactivating pro-apoptotic factors such as Bad and procaspase-9 by phosphorylating them (Cardone et al., 1998; Altomare and Testa, 2005). Indeed, after the treatment with Cuc 1, Akt phosphorylation was down regulated in A549 cells indicating an inhibition of this signaling pathway by Cuc 1 (Fig. 6c). This effect might contribute to apoptosis induction.
CONCLUSION
In conclusion, it can be postulated that Cuc 1 induces apoptosis in A549 cells. This is mediated by G2/M phase cell cycle arrest and actin cytoskeleton disruption. Furthermore, Cuc 1 inhibits the activation of STAT-3 and Akt signaling pathways to down-regulate the expression of Bcl-2. This prompts cytochrome c to be released from the mitochondria to the cytosol which is an essential step for caspases -9 and -3 activation. Therefore, Cuc 1 can be taken to mean a new potent compound with a high anticancer potential due to its cytotoxic and apoptosis inducing effects on malignant lung cells.
ACKNOWLEDGMENTS
The authors acknowledge the financial support received from Brazilian funding agencies: CNPq/MCTI (grant number 472979/2011-6) and PRONEX/FAPESC (grant number 2671/2012-9) as well as the first one for their research fellowships. The study sponsors had no involvement in the design, collection, analysis and interpretation of the data and the decision to submit the manuscript for publication in International Journal of Cancer Research. We also would like to thank Rafael Matielo, Jadel Kratz and Annelise de Carvalho for their proficient editorial assistance.
REFERENCES
- Al Zaid Siddiquee, K. and J. Turkson, 2008. Stat3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res., 18: 254-267.
CrossRef - Altomare, D.A. and J.R. Testa, 2005. Perturbations of the akt signaling pathway in human cancer. Oncogene, 24: 7455-7464.
CrossRef - Budihardjo, I., H. Oliver, M. Lutter, X. Luo and X. Wang, 1999. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol., 15: 269-290.
CrossRefDirect Link - Chan, K.T., K. Li, S.L. Liu, K.H. Chu, M. Toh and W.D. Xie, 2010. Cucurbitacin B inhibits STAT3 and the Raf/MEK/ERK pathway in leukemia cell line K562. Cancer Lett., 289: 46-52.
CrossRef - Chen, J.C., M.H. Chiu, R.L. Nie, G.A. Cordell and S.X. Qiu, 2005. Cucurbitacins and cucurbitane glycosides: Structures and biological activities. Nat. Prod. Rep., 22: 386-399.
CrossRefPubMedDirect Link - Chen, W., A. Leiter, D. Yin, M. Meiring, V.J. Louw and H.P. Koeffler, 2010. Cucurbitacin B inhibits growth, arrests the cell cycle and potentiates antiproliferative efficacy of cisplatin in cutaneous squamous cell carcinoma cell lines. Int. J. Oncol., 37: 737-743.
CrossRef - Haritunians, T., S. Gueller, L. Zhang, R. Badr and D. Yin et al., 2008. Cucurbitacin B induces differentiation, cell cycle arrest, acnd actin cytoskeletal alterations in myeloid leukemia cells. Leuk. Res., 32: 1366-1373.
PubMed - Hengartner, M.O., 2000. The biochemistry of apoptosis. Nature, 407: 770-776.
CrossRefPubMedDirect Link - Ishdorj, G., J.B. Johnston and S.B. Gibson, 2011. Cucurbitacin-I (JSI-124) activates the JNK/c-Jun signaling pathway independent of apoptosis and cell cycle arrest in B Leukemic Cells. BMC Cancer, Vol. 11.
CrossRef - Jayaprakasam, B., N.P. Seeram and M.G. Nair, 2003. Anticancer and antiinflammatory activities of cucurbitacins from Cucurbita andreana. Cancer Lett., 10: 11-16.
CrossRefPubMedDirect Link - Kim, P.K., S.Y. Park, P.P. Koty, Y. Hua, J.D. Luketich and T.R. Billiar, 2003. Fas-associating death domain protein overexpression induces apoptosis in lung cancer cells. J. Thorac. Cardiovasc. Surg., 125: 1336-1342.
CrossRefDirect Link - Lee, D.H., G.B. Iwanski and N.H. Thoennissen, 2010. Cucurbitacin: Ancient compound shedding new light on cancer treatment. Sci. World J., 10: 413-418.
CrossRef - Liu, T., H. Peng, M. Zhang, Y. Deng and Z. Wu, 2010. Cucurbitacin B, a small molecule inhibitor of the Stat3 signaling pathway, enhances the chemosensitivity of laryngeal squamous cell carcinoma cells to cisplatin. Eur. J. Pharmacol., 641: 15-22.
CrossRef - Mosmann, T., 1983. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods, 65: 55-63.
CrossRefPubMedDirect Link - Nicholson, D.W., 2000. From bench to clinic with apoptosis-based therapeutic agents. Nature, 407: 810-816.
CrossRef - Rios, J.L., I. Andujar, J.M. Escandell, R.M. Giner and M.C. Recio, 2012. Cucurbitacins as inducers of cell death and a rich source of potential anticancer compounds. Curr. Pharm. Des., 18: 1663-1676.
PubMedDirect Link - Siegel, R., E. Ward, O. Brawley and A. Jemal, 2011. Cancer statistics, 2011: The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J. Clinicians, 61: 212-236.
CrossRefPubMedDirect Link - Su, Y., G. Li, X. Zhang, J. Gu, C. Zhang, Z. Tian and J. Zhang, 2008. Jsi-124 inhibits glioblastoma multiforme cell proliferation through g(2)/m cell cycle arrest and apoptosis augment. Cancer Biol. Ther., 7: 1243-1249.
PubMedDirect Link - Tannin-Spitz, T., S. Grossman, S. Dovrat, H.E. Gottlieb and M. Bergman, 2007. Growth inhibitory activity of cucurbitacin glucosides isolated from Citrullus colocynthis on human breast cancer cells. Biochem. Pharmacol., 73: 56-67.
CrossRefPubMedDirect Link - Wakimoto, N., D. Yin, J. O'Kelly, T. Haritunians and B. Karlan et al., 2008. Cucurbitacin B has a potent antiproliferative effect on breast cancer cells in vitro and in vivo. Cancer Sci., 99: 1793-1797.
CrossRefDirect Link - Yu, H. and R. Jove, 2004. The STATs of cancer-new molecular targets come of age. Nat. Rev. Cancer, 4: 97-105.
CrossRefDirect Link