
S.A. Sultana
Golden Homes, Tolichowki, Hyderabad, Andhra Pradesh, India
S. Kiranmayee
Stapenhill Medical Centre, Burton-on-Trent, Staffordshire, DE15 9QD, United Kingdom
V.K. Bammidi
Stapenhill Medical Centre, Burton-on-Trent, Staffordshire, DE15 9QD, United Kingdom
A.P. Shaik
Jawaharlal Nehru Institute of Advanced Studies, Buddha Bhavan, MG Road, Secunderabad, Andhra Pradesh, India
K. Jamil
Jawaharlal Nehru Institute of Advanced Studies, Buddha Bhavan, MG Road, Secunderabad, Andhra Pradesh, India
International Journal of Cancer Research
Year: 2011 | Volume: 7 | Issue: 2 | Page No.: 99-113







ABSTRACT
The role of fragile histidine triad (fhit) gene in the etiology of cancer is a relatively recent area of research. The fhit gene has been investigated in most cancers; however, literature is not conclusive regarding its role in the pathophysiology of cancer. Many studies are now focusing on this gene and its potential relationship with cancers. Although, studies have shown an association between infection with Human Papillomavirus (HPV) and cervical neoplasia, evidence also suggests that this infection alone is not sufficient for development of cervical cancer. Other genetic factors like altered tumor suppressor gene activities are also thought to contribute to the carcinogenic process in cervical carcinomas. In this short review, we present the function, the potential role of fhit gene and its protein, influence of fhit gene in various cancers with specific emphasis on cervical cancer has been discussed. In addition, the present article also focuses on the biochemical and molecular nature of FHIT protein.
PDF Abstract XML References Citation
Received: November 06, 2010;
Accepted: November 12, 2010;
Published: April 22, 2011
How to cite this article
S.A. Sultana, S. Kiranmayee, V.K. Bammidi, A.P. Shaik and K. Jamil, 2011. Introduction to the Role of Fragile Histidine Triad (fhit) Gene in Cancer: A Review of Literature with Special Emphasis on Cervical Carcinoma. International Journal of Cancer Research, 7: 99-113.
DOI: 10.3923/ijcr.2011.99.113
URL: https://scialert.net/abstract/?doi=ijcr.2011.99.113
DOI: 10.3923/ijcr.2011.99.113
URL: https://scialert.net/abstract/?doi=ijcr.2011.99.113
INTRODUCTION
Exposure to carcinogens and the associated FHIT inactivation was first observed in lung cancers (Sozzi et al., 1998), suggesting that the alteration of the fhit gene through damage to the associated fragile region by carcinogens may contribute in a large part to the pathophysiology of cancer. The loss of FHIT function was more frequently observed in cancers developing in individuals with constitutional alterations to genes involved in DNA repair (Mori et al., 2001). Epigenetic changes to chromatin, such as DNA methylation and modifications in histone proteins regulate transcription of several tumor suppressor genes (Hsieh and Jones, 2003). Multiple genetic changes, including activation of protooncogenes to oncogenes and epigenetic modification (inactivation) of tumor-suppressor genes are involved in the pathogenesis of cancer (Hsieh and Jones, 2003; Pichiorri et al., 2008). Such genetic changes affect cell survival, cell proliferation and stability of the genome. The 3p14.2 region in chromosome 3 which harbors the fhit gene encompasses the most active common fragile sites of the human genome making the region very sensitive to alterations by DNA damaging agents (Pichiorri et al., 2008).
Aberrant transcripts of the fhit (fragile histidine triad) gene have been reported in human cancers (Ohta et al., 1996; Sozzi et al., 1996; Mao et al., 1996), supporting evidence also indicates the role of FHIT protein in the regulation of apoptosis and cell cycle (Sard et al., 1999). Knockout mice models become highly susceptible to chemical induction of tumors and cells without FHIT protein showed increased resistance to ultra-violet radiation, mitomycin C and ionizing radiation (Ishii et al., 2007). Ishii et al. (2006) suggested that FHIT protects cells from accumulating DNA damages, through modulation of checkpoint proteins Hus1 and phosphoChk1.
Protein biochemistry: FHIT belongs to the histidine triad superfamily (HIT, characterized by the histidine triad motif, HxHxHxx (where, x is a hydrophobic residue)) of nucleotide-binding proteins and is functionally a diadenosine triphosphate hydrolase which under in vitro conditions cleaves diadenosine triphosphate through a magnesium dependent hydrolysis to adenine diphosphate and adenine monophosphate (Barnes et al., 1996). In humans, this enzyme is composed of 147 amino acids (Ohta et al., 1996; Pekarsky et al., 1998) and is involved in purine metabolism. In the first step of enzymatic hydrolysis, it is hypothesized that diadenosine triphosphateand Mg(2+) reacts with the His96 residue of the enzyme to form a covalent FHIT-AMP intermediate releasing Mg-ADP; this intermediate in the second step releases AMP (Barnes et al., 1996; Abend et al., 1999). Barnes et al. (1996) showed that the FHIT substrates, diadenosine triphosphate and diadenosine tetraphosphate are involved in intracellular functions such as regulation of DNA replication and signaling stress responses. Direct evidence of this mechanism was provided by Huang et al. (2004), who showed that FHIT protein mutated at His96 region is completely inactive against Mg-diadenosine diphosphate indicating that conserved residues of the histidine triad are required for activity of the enzyme.
The FHIT protein is homologous to the Aph1 enzyme of Saccharomyces pombe, which has diadenosine triphosphate hydrolase activity, a function that is conserved from yeast to human. Structural studies have shown that binding of the FHIT dimer with two molecules of diadenosine triphoshate, results in highly phosphorylated surfaces, with potential signaling activity (Pace et al., 1998; Pekarsky et al., 2004). The FHIT protein may exist is unphosphorylated, monophosphorylated and diphosphorylated forms.
The 16.8 KDa protein produced by the expression of the fhit gene is phosphorylated at tyrosine 114 residue by Src family proteins. The structure of the FHIT protein along with active sites predicted by thematics is shown in Fig. 1. The FHIT protein is expressed at the highest steady state levels in the kidney and liver.
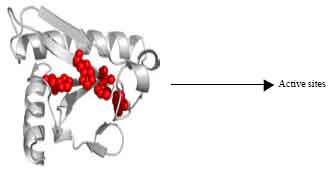 | |
| Fig. 1: | FHIT protein with active sites. Active sites of the FHIT protein as predicted by Thematics. Human fragile histidine triad protein (E.C. 3.6.1.29; PDB: 5FIT). Adapted from: Wei et al. (2007) |
The protein is primarily cytosolic, but is also found in the mitochondria. Restoration of fhit gene expression in cancer cells deficient in this gene causes death by apoptosis, involving the intrinsic caspase pathway, in cancer-derived cells and in tumor xenografts. The tumor-specific loss or reduction of FHIT protein has been detected immunohistochemically in cervical carcinoma (Greenspan et al., 1997).
Interactions of FHIT with other proteins: The biological function of FHIT protein was characterized using the yeast two-hybrid screen by Shi et al. (2000). FHIT was shown to interact with the protein UBE2I, this sequence was found to be identical to that of human ubiquitin-conjugating enzyme 9 (hUBC9). A single amino acid substitution at codon 96 from histidine to asparagine (His_Asn) or three amino acid substitutions (His_Asn) at codons 94, 96 and 98 did not affect this association. The enzymatic activity of FHIT was eliminated by mutations in either of the histidine triad regions indicating the potential role for this protein in controlling cell cycle.
Studies of Weiske et al. (2007) showed that FHIT protein is associated with a lymphoid enhancer-binding factor-1, T-cell factor and beta-catenin complex in human embryonic kidney cells. FHIT was shown to be bound to the C-terminal domain of beta-catenin, a protein which plays a vital role in the Wnt signalling pathway.
Mutations in fhit gene: The fhit (fragile histidine triad) gene spanning more than 1.6 Mb of the genomic DNA is a tumor-suppressor gene composed of 10 exons. The gene encodes a 1.1 kb mRNA. The most common fragile site, FRA3B is located within the fhit gene (Ohta et al., 1996). It has been shown that the degree of chromosomal fragility at this particular site may determine the degree of susceptibility to cancer (Yang et al., 2002). The fhit region also encompasses the break point of the t(3:8) translocation (Fig. 2), identified in familial renal-cell carcinoma. Using transfection experiments in 4 different cell line with homozygous deletions of the fhit gene, Croce et al. (1999) demonstrated the tumor suppressor activity of this gene. The FHIT-expressing transfectants when injected into nude mice resulted in the loss of the ability to form tumors (Siprashvili et al., 1997). Ohta et al. (1996) showed that three 5-prime exons of FHIT are centromeric to the 3p14.2 breakpoint, while the remaining exons are telomeric to this region (Fig. 3). Using FHIT gene knock-out mouse embryonal stem cells, Fong et al. (2000) and Zanesi et al. (2001) have shown that the fhit -/- knockout mice depicted increased susceptibility to spontaneous tumors and high sensitivity to carcinogens. Aberrant fhit gene transcripts have been found in esophageal, stomach and other carcinomas. A pseudogene, with sequences nearly identical to the 51UTR of FHIT, was found to be located on chromosome 1 (www.atlasgeneticsoncology.org/Genes/FHITID192ch3p).
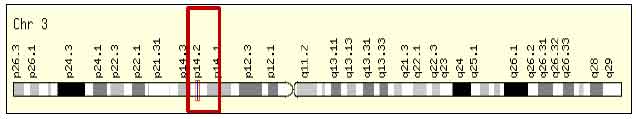 | |
| Fig. 2: | Chromosomal location of fhit gene. The 3p14.2 region in chromosome 3 which harbors the fhit gene. Adapted from: http://www.genecards.org/cgi-bin/carddisp.pl?gene=Fhit |
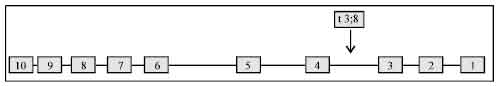 | |
| Fig. 3: | Exons of fhit gene. Three 5-prime exons of FHIT are centromeric to the 3p14.2 breakpoint, while the remaining exons are telomeric to this region. The figure shows the fhit gene genomic locus with exons and the position of t(3;8) translocation. Redrawn based on image from: www.cancerindex.org/clinks3h.htm |
Corbin et al. (2002) suggested the probable presence of multiple hot spots within the FHIT/FRA3B locus. Their experiments involved microcell-mediated chromosome transfer to isolate hybrid cell clones that retain chromosome 3 homologues followed by molecular mapping of the FHIT/FRA3B locus. Their results also suggest that factors other than the DNA sequence alone may be responsible for DNA breaks/gaps.
Implications of fhit gene mutations: Some of the fhit gene polymorphisms that have been most commonly observed in human cancers include the below:
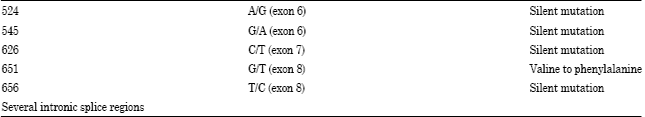 |
| Adapted from: www.atlasgeneticsoncology.org/Genes/FHITID192ch3p |
Homozygous deletions in fhit gene in lung, gastrointestinal, breast and head-and-neck cancers and aberrant fhit transcripts in cancer cell lines have been reported (Ohta et al., 1996; Sozzi et al., 1996; Negrini et al., 1996; Virgilio et al., 1996; Druck et al., 1997). However, aberrant fhit transcripts have been seen in histologically normal tissues (Latil et al., 1998). Although, Fong et al. (1997) have reported that the point mutation of the fhit gene is rare in lung cancer, Yoshino et al. (1998) in contrast found 9 point mutations (19%) in cervical carcinomas and showed that these are somatic mutations rather than rare polymorphisms or germline mutations.
Animal models to investigate the role of fhit gene: After inactivation of the fhit allele, the resultant mice carrying the inactivated fhit allele (+/-) were treated with nitrosomethylbenzylamine. While only 25% of the +/+ mice developed adenoma or papilloma, 100% of fhit deficient mice developed multiple tumors. The visceral and sebaceous tumors, which lacked FHIT protein, were similar to the tumors found in the Muir-Torre familial cancer syndrome (Fong et al., 2000).
Zanesi et al. (2001) suggest that the fhit gene may be a one-hit tumor suppressor gene in some tissues. Dumon et al. (2001) inhibited tumor development by fhit gene transfer using viral vectors suggesting that fhit gene therapy could be a novel clinical approach in cancer. Shiraishi et al. (2001) sequenced >600 kb of the mouse locus and determined the fhit deletion breakpoints in a mouse kidney cancer cell line. Sequence alignment of the murine and human FRA3B sequences showed that this region was stable in evolution. There were also several unusual highly conserved regions.
Role of FHIT in various cancers: The inactivation of fhit gene was shown in a variety of human malignancies indicating its the tumor suppressor function. In approximately 50% of gastrointestinal carcinomas (esohageal, stomach and colon) aberrant fhit transcripts have been identified (Ohta et al., 1996). Huebner and Croce (2003) showed that FHIT is altered in many human tumors caused by environmental carcinogens. The same authors in a previous study (Huebner and Croce, 2001) showed that fhit-negative cancer cells were highly sensitive to the fhit expression. Geurts et al. (1997) reported that FHIT was involved in a translocation-derived fusion with the high-mobility group (non histone chromosomal) protein isoform I-C, the causative gene in a variety of benign tumors. Using sequencing and Southern blot analysis, Rassool et al. (1996) found neither (CGG)n repeats nor other sequences associated with rare fragile sites within the 85 kb contig.
By sequence analysis of fhit locus and 22 associated cancer cell deletion endpoints, Inoue et al. (1997) demonstrated that this locus is a frequent target of homologous recombination resulting in fhit gene internal deletions. Corbin et al. (2002) suggested the possibility of existence of the presence of multiple hot spots within the fhit locus using microcell-mediated chromosome transfer.
Lung cancers: The expression of the FHIT protein and its relevance to the diagnosis and prognosis of lung cancers were studied by Feng et al. (2007) wherein a total loss or marked reduction of expression was seen in 67% of the analyzed cases. This loss or marked reduction of fhit gene expression was lung cancers of smokers. However, the expression of FHIT was not associated with histopathologic grading of tumors and their clinical staging, lymph node metastasis or survival time. The correlation between loss of fhit gene expression with a large number of molecular genetic and clinical parameters in Non-Small-Cell Lung Cancers (NSCLC) were studied using a polyclonal antibody to FHIT protein. A complete loss of cytoplasmic FHIT staining was seen in this immunohistochemical reaction in more than 50% of tumors.
Using reverse transcriptase polymerase chain reaction, Sozzi et al. (1996) analyzed the structure of fhit gene in small cell and non-small cell lung cancers. The authors noted abnormal-sized transcripts in tumors and loss of heterozygosity for microsatellite markers in and regions adjacent to fhit locus. In these tumors, inactivation of the fhit gene occurred loss of 1 allele and altered expression of the remaining allele.
Studies have indicated a role of cigarette smoking in the etiology of lung cancer. Microsatellite alterations within the fhit gene and at an independent locus in chromosome 10 called D10S197 were assessed in lung tumors from heavy smokers and in tumors from never smokers (Sozzi et al., 1997a). Loss of heterozygosity affecting at least one locus of the fhit gene was observed in 80% tumors from the smokers group and only in 22% tumors from non-smokers. While, the loss of fhit in smokers and nonsmokers was statistically significant, no difference in loss of heterozygosity rate was observed at D10S197 locus. An analysis of lung cancer cell lines, small cell lung carcinomas and pairs of non-small cell primary tumors and bronchial mucosa specimen using molecular, genetic and histochemical methods showed concordance between RNA abnormalities and lack of FHIT protein expression in lung tumors and cell lines (Sozzi et al., 1997b). This study also suggested that FHIT protein may be lost at very early stages of lung carcinogenesis. In addition, Stein et al. (2002) showed that active smokers had a significantly higher frequency of fragile site expression, compared to nonsmokers and SCLC patients who stopped smoking. Active tobacco exposure may thus increase the expression of fhit gene.
Head and neck cancers: Virgilio et al. (1996) noted several regions of loss of heterozygosity in head and neck cancers. More than 90% of the analysed head and neck squamous cell carcinoma cell lines showed alterations of at least 1 allele of the fhit gene. Using immunohistochemical analysis Paradiso et al. (2004) hypothesise that decreasing levels of FHIT is directly involved in cancer development.
Thyroid tumors: A retrospective analysis of fhit mRNA transcripts and genomic DNA from thyroid tumors showed the frequent present of truncated fhit transcripts alongwith full-length transcripts (McIver et al., 2000). The pathogenetic role for these aberrant transcripts remains a possibility, but no correlation was found with stage, histological grade or outcome in this study.
Colorectal cancers: Morikawa et al. (2000) reported altered expression of FHIT protein in 47% of colorectal adenomas. The amount of FHIT protein produced was inversely proportional to the degree of dysplasia. Their findings suggest that altered expression of the fhit gene is an early event in the etiology of colorectal cancer. Elnatan et al. (1999) showed that the HIT family of genes were selectively involved in tumorigenesis and also confirmed the early alterations in fhit gene expression. The alteration of the fhit locus and loss of FHIT protein expression were found to be significantly more frequent in sporadic colorectal carcinomas (Mori et al., 2001).
Bladder cancer: Maruyama et al. (2001) investigated the aberrant promoter methylation profile of bladder cancers and correlated their data with clinicopathological findings. The methylation profile was hypothesised to be a potential new biomarker of risk prediction in bladder cancer.
Esophageal cancer: In esophageal cancer, 50% of severe and moderate dysplasias and 33% of mild dysplasias were fhit negative in most of the in situ lesions (Mori et al., 2001). Mimori et al. (2003) showed that microsatellite instability is significantly related to the allelic loss in the fhit region and this was unrelated to the progression of esophageal cancer.
Hepatocellular cancer: Zekri et al. (2005) showed that the fhit gene is a frequent target in hepatitis C virus-associated hepatocellular carcinoma and that alterations affecting this gene occurs as an early event in this type of neoplasm. In addition, studies also showed abnormal apoptosis-proliferation balance indicating an important role of fhit gene expression in the carcinogenesis and development of hepatocellular carcinoma (Nan et al., 2005).
Prostate cancer: A linkage analysis by of 80 candidate genes conducted in prostatic adenocarcinoma showed that involvement of germline variations of FHIT may increase the risk of prostate cancer risk (Larson et al., 2005).
Breast cancer: The protective role of fhit gene was determined by crossing in mice carrying one inactivated fhit allele with mice carrying the rat neu proto-oncogene (Bianchi et al., 2007). All fhit heterozygous mice developed mammary tumors, whereas when both fhit alleles were present, tumor incidence was reduced in 27% of the mice. Their findings suggest a protective role for FHIT in HER2-driven mammary tumors.
The loss of heterozygosity in D3S1300, an fhit intragenic marker with concomitant loss of BRCA1 intragenic marker was reported (Santos et al., 2004). In this study, no correlations were found between loss of heterozygosity with the size of tumor, grade and axillary lymph node metastasis. Guler et al. (2004) observed a strong correlation between FHIT and Wwox expression, a result consistent with the increased susceptibility of fragile sites to DNA damage. The reduced expression of fhit expression was associated with adverse prognostic factors. In an additional study Guler et al. (2009) showed that reduced expression levels of FHIT, Wwox and nuclear AP2gamma have roles in basal-like differentiation in breast cancer. In addition, Wang et al. (2008) have shown that the expression of fhit gene and Wwox and decreases along with progression from normal cells to cancer.
Gastric cancer: The absence of FHIT protein correlated with tumor stage and histologic grade in gastric cancers (Capuzzi et al., 2000). Lee et al. (2001a, b) showed that a higher frequency of aberrant transcripts in gastric carcinomas. Along with this, they observed a significant rate of loss of heterozygosity indicating the important role of fhit gene in etiology of gastric carcinogenesis. A study by Zheng et al. (2007) showed that in gastric cancer, the expression of fhit and pten were lower compared to the levels in normal mucosal cells. A negative association was found with the extent of lymphatic invasion, lymph node metastasis, liver metastasis and staging. However, a positive association was found between fhit and pten gene expression.
Periocular sebaceous gland carcinoma: Holbach et al. (2002) showed that the inactivation of the fhit gene or inactivation of the mismatch-repair system may contribute to the development of periocular sebaceous gland carcinoma in Muir-Torre syndrome.
Renal carcinoma: Gemmill et al. (1998) showed that the reciprocal t (3;8) translocation was associated with multifocal clear cell renal carcinoma. Such balanced and constitutional translocation was also reported in bilateral clear cell renal cell carcinoma (Poland et al., 2007), these translocations disrupt the trc8 and fhit genes caused increased susceptibility to bilateral renal cell carcinoma. Strefford et al. (2005) reported genomic imbalances and rearrangements in renal cell carcinoma cell lines. The frequent loss or decrease in FHIT protein expression was also observed by Gayrard et al. (2008) in addition fhit inactivation was found to play a major role in renal cell tumorigenesis by Velickovic et al. (2001) and Sukosd et al. (2003).
Role of FHIT gene in cervical cancers: Loss of FHIT expression, was found to be reported commonly in patients with stage IA1 to IB2 cervical squamous cell carcinoma, this being significantly more common in cervical cancers of smokers (Holschneider et al., 2005). Abnormal protein expression fhit gene has been reported to play a pivotal role in cervical cancers (www.cancerindex.org/clinks3h.htm), a common type of malignancy accounting for about 6% of all cancers found in women (Parkin et al., 1999). Cervical cancer remains a major source of cancer-related morbidity and death for women throughout the world (Bosch et al., 1995). The Human Papilloma Viruses (HPV) are the principal cause of cervical cancer and HPV DNA has been found in more than 95% of carcinoma cases. HPV are DNA viruses and affect the nucleus and the cytoplasm of the infected cells. The DNA of these viruses may or may not be intergrated in the epithelial cell nuclei. However, morphological changes occur when HPV DNA is integrated into the epithelial cell nuclear DNA. The HPV lesions are typically dysplastic and atypical and may be associated with chromosome and ploidy alterations (Geradts et al., 2000). Infection with certain high risk Human Papilloma Virus (HPV) types is associated with cervical cancer probably by inactivation of p53 and pRB through interactions with the HPV E6 and E7 proteins, respectively (Alani and Munger, 1998). HPV infections are generally transient, but a small percentage of women develop cervical cancer (Zur Hausen, 1990; Evander et al., 1995).
Nevertheless, HPV infection alone may not be sufficient for the development of cervical cancer since this does not explain the additional events that are necessary for some infections to become chronic and undergo malignant transformation (Neyaz et al., 2010). Genomic rearrangements, aberrant mRNA transcripts and decreased or completed absence of FHIT protein have been reported in cervical carcinomas (Noronha et al., 1999; Lopez-Beltran and Munoz, 1995). These aberrations may have a predictive role in the malignant transformation of lesions in the cervix.
Becker et al. (2002) showed fragility of fhit gene in cervical cancer. The fhit gene expression on a panel of cervical tumor-derived cell lines showed aberrant regulation. In squamous-cell carcinomas of the uterine cervix, 43% of tumors were found with aberrant transcripts and 32% tumors with point mutations (Yoshino et al., 2000). Alterations in fhit were significantly associated with cervical carcinogenesis. Further, this study analysed the alteration of fhit gene in various grades of cervical intra-epithelial neoplasias and invasive cervical carcinomas compared to normal cervical epithelium. A strong association of altered FHIT protein expression with the disruption of normal fhit transcript was observed. There was no correlation between fhit inactivation and HPV infection. The fhit-gene inactivation was shown to be a late event in cervical carcinogenesis. In addition, homozygous deletions of fhit in cervical cancer and cervical carcinoma cell lines have been reported (Muller et al., 1998). Aberrant fhit transcripts were seen only in cervical tumor tissue and not in normal cervical tissue (Greenspan et al., 1997; Muller et al., 1998; Nakagawa et al., 1999; Segawa et al., 1999; Yoshino et al., 2000), however, some studies have reported the same levels of RNA expression pattern in both tumors and normal cervical tissue (Chu et al., 1998; Su et al., 1998; Yoshino et al., 2000).
A reduced FHIT protein level compared to normal cervical epithelium has been reported (Greenspan et al., 1997; Segawa et al., 1999; Birrer et al., 1999; Yoshino et al., 2000). 61% of squamous carcinomas and 40% of adenocarcinomas of the cervix showed abnormal fhit expression (Birrer et al., 1999). fhit expression was abnormal in both glandular and squamous cervical cancers. Abnormal fhit expression was also detected in some preneoplastic lesions of the ectocervix. Alterations in fhit expression may be an important marker of early progression in cervical cancer.
Connolly et al. (2000) suggest that loss of fhit expression could serve as a useful marker of high-grade preinvasive lesions that have an increased likelihood to progress to invasive carcinoma. Baykal et al. (2003) showed that fhit gene was lower in 53% of the cervical carcinomas. None of the clinicopathologic prognostic parameters investigated in this study showed a correlation with FHIT expression. FHIT was thus shown to play a carcinogenic role in tumoral progression but not in the tumoral development.
Bahnassy et al. (2006) reported that aberrations in the fhit gene frequent in HPV-associated cervical carcinoma can be used as predictors of tumor recurrences. In addition to this, a decreased or complete absence of FHIT protein expression was found in 65% cases. The difference between the expression level of the fhit gene and the FHIT protein was reported by Birrer et al. (1999) and Bahnassy et al. (2006). Genomic rearrangements, altered mRNA transcripts and absence or reduction of the FHIT protein have all been reported in numerous epithelial tumors including cervical carcinomas (Lopez-Beltran and Munoz, 1995; Munoz et al., 1995; Noronha et al., 1999). These aberrations may play a predictive role in the identification of the malignant potential of high-grade squamous intraepithelial lesions of the uterine cervix (Schiffman et al., 1993).
Kannan et al. (2000) identified 2 different mutations in oral cancer (caused by chewing tobacco) and cervical cancer (caused by HPV) infection. These mutations were at the second nucleotide 3' to the termination codon (TGA) in exon 9 and at the ninth nucleotide upstream to the beginning of exon 9. In addition to this, the authors also reported a single nucleotide fhit gene polymorphism due to T/A replacement at 17 nucleotides upstream to exon 9.
Neyaz et al. (2008) analysed cervical cancer tissue biopsies of various clinical stages and histological grading. Aberrant promoter methylation of the fhit gene was found in 28.3% of subjects and was significantly (p<0.01) associated with the cervical cancer compared with controls. Neyaz et al. (2010) also attempted to study the role of point mutation in fhit gene in HPV mediated cervical cancer and identified a novel mutation at codon 98 from with replacement of the amino acid His by Arg in cervical cancer. The authors suggest that the His to Arg substitution in the substrate-binding domain may generate catalytically inactive protein with consequent loss of tumor suppressor activity. Promoter hypermehylation and loss of heterozygosity of the fhit was investigated in cervical cancer (Choi et al., 2007). While, promoter hypermethylation was detected in 24% of tumors compared to noncancerous tissues, no correlation was observed between loss of heterozygosity and promoter hypermethylation for fhit gene.
CONCLUSIONS
From the reviewed studies it can be said that, in addition to HPV infection, mutations in fhit gene may be critical for the development and progression of cervical cancer. In addition, from the literature, it is also clear that fhit gene might play an important role in the etiology of several other cancers, although supporting literature is limited. Host factors like altered tumor suppressor gene and activation of protooncogenes to oncogenes that contribute to the carcinogenic process in cervical carcinomas and other cancers need to be further evaluated to understand the specific role of this protein. The present review article focused on the biochemical and molecular nature of FHIT protein the role of this gene as evident from literature in various cancers with special emphasis on cervical cancer. However, to better understand this protein, further studies in larger population sizes and sub-divided into groups based on stages of cancers in necessary.
The data presented here are from a non-homogeneous collection of information available about fhit gene. This is indeed a potential limitation of the study. However, the article tries to give a clear picture of the important role the fhit gene plays in the pathophysiology of cancers. The article therefore focused on the summary of the gene, its protein, expression and its role giving an overall understanding of this gene and its protein.
REFERENCES
- Sozzi, G., U. Pastorino, L. Moiraghi, E. Tagliabue and F. Pezzella et al., 1998. Loss of FHIT function in lung cancer and preinvasive bronchial lesions. Cancer Res., 58: 5032-5037.
Direct Link - Mori, M., K. Mimori, T. Masuda, K. Yoshinaga, K. Yamashita, A. Matsuyama and H. Inoue, 2001. Absence of Msh2 protein expression is associated with alteration in the FHIT locus and Fhit protein expression in colorectal carcinoma. Cancer Res., 61: 7379-7382.
PubMed - Ohta, M., H. Inoue, M.G. Cotticelli, K. Kastury and R. Baffa et al., 1996. The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma-associated t(3;8) breakpoint is abnormal in digestive tract cancers. Cell, 84: 587-597.
PubMed - Sozzi, G., M.L. Veronese, M. Negrini, R. Baffa and M.G. Cotticelli et al., 1996. The FHIT gene 3p14.2 is abnormal in lung cancer. Cell, 85: 17-26.
CrossRef - Sard, L., P. Accornero, S. Tornielli, D. Delia and G. Bunone et al., 1999. The tumor-suppressor gene FHIT is involved in the regulation of apoptosis and in cell cycle control. Proc. Nat. Acad. Sci. USA., 96: 8489-8492.
Direct Link - Ishii, H., Y. Wang and K. Huebner, 2007. A fhit-ing role in the DNA damage checkpoint response. Cell Cycle, 6: 1044-1048.
PubMed - Barnes, L.D., P.N. Garrison, Z. Siprashvili, A. Guranowski and A.K. Robinson et al., 1996. Fhit, a putative tumor suppressor in humans is a dinucleoside 5`,5```-P1,P3-triphosphate hydrolase. Biochemistry, 35: 11529-11535.
PubMed - Pekarsky, Y., T. Druck, M.G. Cotticelli, M. Ohta and J. Shou et al., 1998. The murine Fhit locus: Isolation, characterization and expression in normal and tumor cells. Cancer Res., 58: 3401-3408.
PubMed - Abend, A., P.N. Garrison, L.D. Barnes and P.A. Frey, 1999. Stereochemical retention of the configuration in the action of Fhit on phosphorus-chiral substrates. Biochemistry, 38: 3668-3676.
PubMed - Huang, K., A. Arabshahi, Y. Wei and P.A. Frey, 2004. The mechanism of action of the fragile histidine triad, Fhit: Isolation of a covalent adenylyl enzyme and chemical rescue of H96G-Fhit. Biochemistry, 43: 7637-7642.
PubMed - Pace, H.C., P.N. Garrison, A.K. Robinson, L.D. Barnes and A. Draganescu et al., 1998. Genetic, biochemical and crystallographic characterization of fhit-substrate complexes as the active signaling form of Fhit. Proc. Nat. Acad. Sci. USA., 95: 5484-5489.
PubMed - Pekarsky, Y., P.N. Garrison, A. Palamarchuk, N. Zanesi and R.I. Aqeilan et al., 2004. Fhit is a physiological target of the protein kinase Src. Proc. Nat. Acad. Sci. USA., 101: 3775-3779.
PubMed - Greenspan, D.L., D.C. Connolly, R. Wu, R.Y. Lei and J.T. Vogelstein et al., 1997. Loss of FHIT expression in cervical carcinoma cell lines and primary tumors. Cancer Res., 57: 4692-4698.
PubMed - Wei, Y., J. Ko, L.F. Murga and M.J. Ondrechen, 2007. Selective prediction of interaction sites in protein structures with thematics. BMC Bioinformatics, 8: 119-119.
CrossRef - Shi, Y., M. Zou, N.R. Farid and M.C. Paterson, 2000. Association of FHIT (fragile histidine triad), a candidate tumour suppressor gene with the ubiquitin-conjugating enzyme hUBC9. Biochem. J., 352: 443-448.
PubMed - Weiske, J., K.F. Albring and O. Huber, 2007. The tumor suppressor fhit acts as a repressor of beta-catenin transcriptional activity. Proc. Nat. Acad. Sci. USA., 104: 20344-20349.
PubMed - Yang, Q., M. Nakamura, Y. Nakamura, G. Yoshimura and T. Suzuma et al., 2002. Two-hit inactivation of FHIT by loss of heterozygosity and hypermethylation in breast cancer. Clin. Cancer Res., 8: 2890-2893.
PubMed - Croce, C.M., G. Sozzi and K. Huebner, 1999. Role of FHIT in human cancer. J. Clin. Oncol., 17: 1618-1624.
PubMed - Siprashvili, Z., G. Sozzi, L.D. Barnes, P. McCue and A.K. Robinson et al., 1997. Replacement of Fhit in cancer cells suppresses tumorigenicity. Proc. Natl. Acad. Sci., 94: 13771-13776.
Direct Link - Fong, L.Y., V. Fidanza, N. Zanesi, L.F. Lock and L.D. Siracusa et al., 2000. Muir-torre-like syndrome in fhit-deficient mice. Proc. Nat. Acad. Sci. USA., 97: 4742-4747.
PubMed - Zanesi, N., V. Fidanza, L.Y. Fong, R. Mancini and T. Druck et al., 2001. The tumor spectrum in FHIT-deficient mice. Proc. Nat. Acad. Sci. USA., 98: 10250-10255.
PubMed - Corbin, S., M.E. Neilly, R. Espinosa, E.M. Davis, T.W. McKeithan and M.M. Le Beau, 2002. Identification of unstable sequences within the common fragile site at 3p14.2: Implications for the mechanism of deletions within fragile histidine triad gene/common fragile site at 3p14.2 in tumors. Cancer Res., 62: 3477-3484.
- Negrini, M., C. Monaco, I. Vorechovsky, M. Ohta and T. Druck et al., 1996. The FHIT gene at 3p14.2 is abnormal in breast carcinomas. Cancer Res., 56: 3173-3179.
PubMed - Virgilio, L., M. Shuster, S. Gollin, M.L. Veronese and M. Ohta, 1996. FHIT gene alterations in head and neck squamous cell carcinomas. Proc. Natl. Acad. Sci., 93: 9770-9775.
Direct Link - Druck, T., P. Hadaczek, T.B. Fu, M. Ohta and Z. Siprashvili et al., 1997. Structure and expression of the human FHIT gene in normal and tumor cells. Cancer Res., 57: 504-512.
PubMed - Latil, A., I. Bieche, G. Fournier, O. Cussenot, S. Pesche and R. Lidereau, 1998. Molecular analysis of the FHIT gene in human prostate cancer. Oncogene, 16: 1863-1868.
PubMed - Fong, K.M., E.J. Biesterveld, A. Virmani, I. Wistuba and Y. Sekido et al., 1997. FHIT and FRA3B 3p14.2 allele loss are common in lung cancer and preneoplastic bronchial lesions and are associated with cancer-related FHIT cDNA splicing aberrations. Cancer Res., 57: 2256-2267.
PubMed - Yoshino, K., T. Enomoto, T. Nakamura, R. Nakashima, H. Wada, J. Saitoh, K. Noda and Y. Murata, 1998. Aberrant FHIT transcripts in squamous cell carcinoma of the uterine cervix. Int. J. Cancer, 76: 176-181.
PubMed - Dumon, K.R., H. Ishii, L.Y. Fong, N. Zanesi and V. Fidanza et al., 2001. FHIT gene therapy prevents tumor development in Fhit-deficient mice. Proc. Nat. Acad. Sci. USA., 98: 3346-3351.
CrossRef - Shiraishi, T., T. Druck, K. Mimori, J. Flomenberg and L. Berk et al., 2001. Sequence conservation at human and mouse orthologous common fragile regions, FRA3B/FHIT and Fra14A2/Fhit. Proc. Nat. Acad. Sci. USA., 98: 5722-5727.
PubMed - Huebner, K. and C.M. Croce, 2001. FRA3B and other common fragile sites: The weakest links. Nat. Rev. Cancer, 1: 214-221.
PubMed - Geurts, J.M., E.F. Schoenmakers, E. Roijer, G. Stenman and W.J. van de Ven, 1997. Expression of reciprocal hybrid transcripts of HMGIC and FHIT in a pleomorphic adenoma of the parotid gland. Cancer Res., 57: 13-17.
PubMed - Rassool, F.V., M.M. le Beau, M.L. Shen, M.E. Neilly and R. Espinosa et al., 1996. Direct cloning of DNA sequences from the common fragile site region at chromosome band 3p14.2. Genomics, 35: 109-117.
PubMed - Inoue, H., H. Ishii, H. Alder, E. Snyder, T. Druck, K. Huebner and C.M. Croce, 1997. Sequence of the FRA3B common fragile region: Implications for the mechanism of FHIT deletion. Proc. Nat. Acad. Sci. USA., 94: 14584-14589.
PubMed - Feng, X., L. Li, Y. Gao, J. Zhang and J. Ying et al., 2007. Fhit protein expression in lung cancer studied by high-throughput tissue microarray. Bull. Cancer, 94: E8-11.
PubMed - Sozzi, G., L. Sard, L. de Gregorio, A. Marchetti and K. Musso et al., 1997. Association between cigarette smoking and FHIT gene alterations in lung cancer. Cancer Res., 57: 2121-2123.
PubMed - Sozzi, G., S. Tornielli, E. Tagliabue, L. Sard and F. Pezzella et al., 1997. Absence of Fhit protein in primary lung tumors and cell lines with FHIT gene abnormalities. Cancer Res., 57: 5207-5212.
PubMed - Stein, C.K., T.W. Glover, J.L. Palmer and B.S. Glisson, 2002. Direct correlation between FRA3B expression and cigarette smoking. Genes Chromosomes Cancer, 34: 333-340.
PubMed - Paradiso, A., G. Ranieri, B. Stea, A. Zito and I. Zehbe et al., 2004. Altered p16INK4a and Fhit expression in carcinogenesis and progression of human oral cancer. Int. J. Oncol., 24: 249-255.
PubMed - McIver, B., S.K. Grebe, L. Wang, I.D. Hay and A. Yokomizo et al., 2000. FHIT and TSG101 in thyroid tumours: Aberrant transcripts reflect rare abnormal RNA processing events of uncertain pathogenetic or clinical significance. Clin. Endocrinol., 52: 749-757.
PubMed - Morikawa, H., Y. Nakagawa, K. Hashimoto, M. Niki and Y. Egashira et al., 2000. Frequent altered expression of fragile histidine triad protein in human colorectal adenomas. Biochem. Biophys. Res. Commun., 278: 205-210.
PubMed - Elnatan, J., D. Murphy, H.S. Goh and D.R. Smith, 1999. HIT family genes: FHIT but not PKCI-1/HINT produces altered transcripts in colorectal cancer. Br. J. Cancer, 81: 874-880.
PubMed - Maruyama, R., S. Toyooka, K.O. Toyooka, K. Harada and A.K. Virmani et al., 2001. Aberrant promoter methylation profile of bladder cancer and its relationship to clinicopathological features. Cancer Res., 61: 8659-8663.
PubMed - Mimori, K., H. Inoue, T. Shiraishi, A. Matsuyama, K. Mafune, Y. Tanaka and M. Mori, 2003. Microsatellite instability is often observed in esophageal carcinoma patients with allelic loss in the FHIT/FRA3B locus. Oncology, 64: 275-279.
PubMed - Zekri, A.R., A.A. Bahnassy, M. Hafez, A.M. El-Shehaby, G.M. Sherif, H.M. Khaled and N. Zakhary, 2005. Alterations of the fragile histidine triad gene in hepatitis C virus-associated hepatocellular carcinoma. J. Gastroenterol. Hepatol., 20: 87-94.
CrossRef - Nan, K.J., Z.P. Ruan, Z. Jing, H.X. Qin, H.Y. Wang, H. Guo and R. Xu, 2005. Expression of fragile histidine triad in primary hepatocellular carcinoma and its relation with cell proliferation and apoptosis. World J. Gastroenterol., 11: 228-231.
PubMed - Larson, G.P., Y. Ding, L.S. Cheng, C. Lundberg and V. Gagalang et al., 2005. Genetic linkage of prostate cancer risk to the chromosome 3 region bearing FHIT. Cancer Res., 65: 805-814.
PubMed - Bianchi, F., E. Tagliabue, S. Menard and M. Campiglio, 2007. Fhit expression protects against HER2-driven breast tumor development: unraveling the molecular interconnections. Cell Cycle, 6: 643-646.
PubMed - Santos, S.C., L.R. Cavalli, I.J. Cavalli, R.S. Lima, B.R. Haddad and E.M. Ribeiro, 2004. Loss of heterozygosity of the BRCA1 and FHIT genes in patients with sporadic breast cancer from Southern Brazil. J. Clin. Pathol., 57: 374-377.
PubMed - Guler, G., A. Uner, N. Guler, S.Y. Han and D. Iliopoulos et al., 2004. The fragile genes FHIT and WWOX are inactivated coordinately in invasive breast carcinoma. Cancer, 100: 1605-1614.
PubMed - Guler, G., K. Huebner, C. Himmetoglu, R.E. Jimenez and S. Costinean et al., 2009. Fragile histidine triad protein, WW domain-containing oxidoreductase protein Wwox and activator protein 2gamma expression levels correlate with basal phenotype in breast cancer. Cancer, 115: 899-908.
PubMed - Wang, T.T., E.E. Frezza, R. Ma, S.Y. Hu and C.Z. Liu et al., 2008. Loss expression of active fragile sites genes associated with the severity of breast epithelial abnormalities. Chin. Med. J., 121: 1969-1974.
PubMed - Capuzzi, D., E. Santoro, W.W. Hauck, A.J. Kovatich and F.E. Rosato et al., 2000. FHIT expression in gastric adenocarcinoma: correlation with disease stage and survival. Cancer, 88: 24-34.
CrossRefDirect Link - Lee, S.H., C.J. Kim, H.K. Park, J.W. Koh, M.H. Cho, M.J. Baek and M.S. Lee, 2001. Characterization of aberrant FHIT transcripts in gastric adenocarcinomas. Exp. Mol. Med., 33: 124-130.
PubMed - Lee, S.H., W.H. Kim, H.K. Kim, K.M. Woo and H.S. Nam et al., 2001. Altered expression of the fragile histidine triad gene in primary gastric adenocarcinomas. Biochem. Biophys. Res. Commun., 284: 850-855.
PubMed - Zheng, H., H. Takahashi, Y. Murai, Z. Cui, K. Nomoto, K. Tsuneyama and Y. Takano, 2007. Low expression of FHIT and PTEN correlates with malignancy of gastric carcinomas: Tissue-array findings. Applied Immunohistochem. Mol. Morphol., 15: 432-440.
PubMed - Holbach, L.M., A. von Moller, C. Decker, A.G. Junemann, C. Rummelt-Hofmann and W.G. Ballhausen, 2002. Loss of fragile histidine triad (FHIT) expression and microsatellite instability in periocular sebaceous gland carcinoma in patients with Muir-Torre syndrome. Am. J. Ophthalmol., 134: 147-148.
Direct Link - Gemmill, R.M., J.D. West, F. Boldog, N. Tanaka and L.J. Robinson et al., 1998. The hereditary renal cell carcinoma 3;8 translocation fuses FHIT to a patched-related gene, TRC8. Proc. Nat. Acad. Sci. USA., 95: 9572-9577.
PubMed - Poland, K.S., M. Azim, M. Folsom, R. Goldfarb and R. Naeem et al., 2007. A constitutional balanced t(3;8)(p14;q24.1) translocation results in disruption of the TRC8 gene and predisposition to clear cell renal cell carcinoma. Genes Chromosomes Cancer, 46: 805-812.
PubMed - Strefford, J.C., I. Stasevich, T.M. Lane, Y.J. Lu, T. Oliver and B.D. Young, 2005. A combination of molecular cytogenetic analyses reveals complex genetic alterations in conventional renal cell carcinoma. Cancer Genet. Cytogenet., 159: 1-9.
PubMed - Gayrard, N., V. Cacheux, F. Iborra, G. Mourad and A. Argiles, 2008. Cytogenetic studies of 24 renal epithelial tumors with von Hippel-Lindau and fragile histidine triad protein expression correlation. Arch. Pathol. Lab. Med., 132: 965-973.
PubMed - Velickovic, M., B. Delahunt, S. Storkel and S.K. Grebem, 2001. VHL and FHIT locus loss of heterozygosity is common in all renal cancer morphotypes but differs in pattern and prognostic significance. Cancer Res., 61: 4815-4819.
PubMed - Sukosd, F., N. Kuroda, T. Beothe, A.P. Kaur and G. Kovacs, 2003. Deletion of chromosome 3p14.2-p25 involving the VHL and FHIT genes in conventional renal cell carcinoma. Cancer Res., 63: 455-457.
PubMed - Holschneider, C.H., R.L. Baldwin, K. Tumber, C. Aoyama and B.Y. Karlan, 2005. The fragile histidine triad gene: A molecular link between cigarette smoking and cervical cancer. Clin. Cancer Res., 11: 5756-5763.
PubMed - Parkin, D.M., P. Pisani and J. Ferlay, 1999. Estimates of the worldwide incidence of 25 major cancers in 1990. Int. J. Cancer, 80: 827-841.
CrossRefPubMedDirect Link - Bosch, F.X., M.M. Manos, N. Muaoz, M. Sherman and A.M. Jansen et al., 1995. Prevalence of human papillomavirus in cervical cancer: A worldwide perspective. International biological study on cervical cancer (IBSCC) Study Group. J. Nat. Cancer Inst., 87: 796-802.
PubMed - Geradts, J., K.M. Fong, P.V. Zimmerman and J.D. Minna, 2000. Loss of fhit expression in non-small-cell lung cancer: Correlation with molecular genetic abnormalities and clinicopathological features. Br. J. Cancer, 82: 1191-1197.
PubMed - Alani, R.M. and K. Munger, 1998. Human papillomaviruses and associated malignancies. J. Clin. Oncol., 16: 330-337.
PubMed - Zur Hausen, H., 1990. The role of papillomaviruses in anogenital cancer. Scand. J. Infect. Dis. Suppl., 69: 107-111.
PubMed - Evander, M., K. Edlund, A. Gustafsson, M. Jonsson, R. Karlsson, E. Rylander and G. Wadell, 1995. Human papillomavirus infection is transient in young women: a population-based cohort study. J. Infect. Dis., 171: 1026-1030.
PubMed - Neyaz, M.K., S. Hussain, M.I. Hassan, B.C. Das, S.A. Husain and M. Bharadwaj, 2010. Novel missense mutation in FHIT gene: Interpreting the effect in HPV-mediated cervical cancer in Indian women. Mol. Cell. Biochem., 335: 53-58.
PubMed - Noronha, V., W. Mello, L. Villa, A. Brito and R. Macedo et al., 1999. Human papillomavirus associated with uterine cervix lesions. Rev. Soc. Bras. Med. Trop., 32: 235-340.
PubMed - Lopez-Beltran, A. and E. Munoz, 1995. Transitional cell carcinoma of the bladder: Low incidence of human papillomavirus DNA detected by the polymerase chain reaction and in situ hybridization. Histopathology, 26: 565-569.
PubMed - Becker, N.A., E.C. Thorland, S.R. Denison, L.A. Phillips and D.I. Smith, 2002. Evidence that instability within the FRA3B region extends four megabases. Oncogene, 21: 8713-8722.
Direct Link - Yoshino, K., T. Enomoto, T. Nakamura, H. Sun and K. Ozaki et al., 2000. FHIT alterations in cancerous and non-cancerous cervical epithelium. Int. J. Cancer, 85: 6-13.
PubMed - Muller, C.Y., J.D. O`Boyle, K.M. Fong, Wistuba and E. Biesterveld et al., 1998. Abnormalities of fragile histidine triad genomic and complementary DNAs in cervical cancer: Association with human papillomavirus type. J. Nat. Cancer Inst., 90: 433-439.
PubMed - Nakagawa, S., H. Yoshikawa, M. Kimura, K. Kawana and K. Matsumoto et al., 1999. A possible involvement of aberrant expression of the FHIT gene in the carcinogenesis of squamous cell carcinoma of the uterine cervix. Br. J. Cancer, 79: 589-594.
CrossRef - Segawa, T., T. Sasagawa, H. Yamazaki, J. Sakaike, H. Ishikawa and M. Inoue, 1999. Fragile histidine triad transcription abnormalities and human papillomavirus E6-E7 mRNA expression in the development of cervical carcinoma. Cancer, 85: 2001-2010.
PubMed - Chu, T.Y., C.Y. Shen, Y.S. Chiou, J.J. Lu, C.L. Perng, M.S. Yu and H.S. Liu, 1998. HPV-associated cervical cancers show frequent allelic loss at 3p14 but no apparent aberration of FHIT mRNA. Int. J. Cancer, 75: 199-204.
PubMed - Su, T.H., J.C. Wang, H.H. Tseng, C.P. Chang, T.A. Chang, H.J. Wei and J.G. Chang, 1998. Analysis of FHIT transcripts in cervical and endometrial cancers. Int. J. Cancer, 76: 216-222.
PubMed - Connolly, D.C., D.L. Greenspan, R. Wu, X. Ren and R.L. Dunn et al., 2000. Loss of fhit expression in invasive cervical carcinomas and intraepithelial lesions associated with invasive disease. Clin. Cancer Res., 6: 3505-3510.
PubMed - Baykal, C., A. Ayhan, A. Al, K. Yuce and A. Ayhan, 2003. No relationship is indicated between FHIT expression and clinicopathologic prognostic parameters in early stage cervical carcinoma. Int. J. Gynecol. Cancer, 13: 192-196.
PubMed - Munoz, N., I. Kato, F.X. Bosch, S. de Sanjose and V.A. Sundquist et al., 1995. Cervical cancer and herpes simplex virus type 2: case-control studies in Spain and Colombia, with special reference to immunoglobulin-G sub-classes. Int. J. Cancer, 60: 438-442.
PubMed - Schiffman, M.H., H.M. Bauer, R.N. Hoover, A.G. Glass and D.M. Cadell et al., 1993. Epidemiologic evidence showing that human papillomavirus infection causes most cervical intraepithelial neoplasia. J. Nat. Cancer Inst., 85: 958-964.
PubMed - Kannan, K., A.K. Munirajan, V. Bhuvarahamurthy, B.K. Mohanprasad, P. Shankar, N. Tsuchida and G. Shanmugam, 2000. FHIT gene mutations and single nucleotide polymorphism in Indian oral and cervical squamous cell carcinomas. Oral. Oncol., 36: 189-193.
PubMed - Neyaz, M.K., R.S. Kumar, S. Hussain, S.H. Naqvi and I. Kohaar et al., 2008. Effect of aberrant promoter methylation of FHIT and RASSF1A genes on susceptibility to cervical cancer in a North Indian population. Biomarkers, 13: 597-606.
PubMed - Choi, C.H., K.M. Lee, J.J. Choi, T.J. Kim and W.Y. Kim et al., 2007. Hypermethylation and loss of heterozygosity of tumor suppressor genes on chromosome 3p in cervical cancer. Cancer Lett., 255: 26-33.
PubMed