
Ajay Mahajan
Department of Periodontics, H.P.G.D.C., Shimla, Himachal Pradesh, India
Bindiya Kumari
Department of Periodontics, H.P.G.D.C., Shimla, Himachal Pradesh, India
Suneet Karol
Department of Periodontics, H.P.G.D.C., Shimla, Himachal Pradesh, India
European Journal of Dentistry and Medicine
Year: 2012 | Volume: 4 | Issue: 3 | Page No.: 38-44







ABSTRACT
Tissue engineering has been defined as the application of principles and methods of engineering and life science towards fundamental understanding of structure function relationship in normal and pathological tissue and development of biological substitute to restore maintain or improve tissue functions. In recent times there’s much hype about the use of tissue engineering techniques in medicine and dentistry. The present study focused on the practical implications of using the tissue engineering techniques in medical and dental treatments and critically evaluates the ground reality behind the approach.
PDF Abstract XML References Citation
Received: August 20, 2012;
Accepted: October 06, 2012;
Published: November 08, 2012
How to cite this article
Ajay Mahajan, Bindiya Kumari and Suneet Karol, 2012. Tissue Engineering: Why So Much Hype?. European Journal of Dentistry and Medicine, 4: 38-44.
DOI: 10.3923/ejdm.2012.38.44
URL: https://scialert.net/abstract/?doi=ejdm.2012.38.44
DOI: 10.3923/ejdm.2012.38.44
URL: https://scialert.net/abstract/?doi=ejdm.2012.38.44
INTRODUCTION
Advances in tissue engineering life sciences over the past twenty years have lead to therapies for replacing, repairing, restoring or regenerating human tissue and organ function and tissue engineering plays the most important role in regenerating the tissue (Hellman et al., 1998). Tissue engineering has been defined as the application of principles and methods of engineering and life science towards fundamental understanding of structure function relationship in normal and pathological tissue and development of biological substitute to restore maintain or improve tissue functions (Lee and Schrank, 2010). There are four basic components which are required for tissue engineering:
| • | A source of stem cells which can divide into the required tissue/organ |
| • | A biochemical mediator (Growth factors) which promotes the growth of stem cells |
| • | A scaffold on which the cells can grow |
| • | Extracellular matrix (ECM) |
When all the three components i.e., the stem cells, biological-mediator and scaffold are used appropriately in combination tissue engineering product is formed which can be used to repair or replace portions of or whole tissues. A number of criterions must be satisfied in order to achieve effective, long-lasting repair of damaged tissues:
| • | An adequate number of cells must be produced to fill the defect |
| • | Cells must differentiate into desired phenotypes |
| • | Cells must adopt appropriate three-dimensional structural scaffold and produce ECM |
| • | Produced cells must be structurally and mechanically compliant with the native cell |
| • | Cells must successfully be able to integrate with native cells and should not evoke any Immunological reaction |
| • | There should be no associated biological risks |
Recent advancements in tissue engineering techniques have created a plethora of tissue engineered products which are now available commercially. A significant portion of this effort has been translated to actual therapies, especially in the areas of skin replacement and to a lesser extent, cartilage repair. A good amount of thoughtful work has also yielded prototypes of other tissue substitutes such as nerve conduits, blood vessels, liver and even heart (Berthiaume et al., 2011).
In the field of dentistry a range of tissue engineered products are available ranging from tissue engineered teeth (Galler and D’Souza, 2011) to oral mucosa (Moharamzadeh et al., 2007). In addition acellular dermal matrix graft has also been utilized for treatment of gingival recessions (Mahajan, 2010). The role of periosteum as a rich source of stem cells (Mahajan, 2011) and the immense regenerative capacity of grafts derived from periosteum has also been highlighted in recent studies (Mahajan, 2009; Mahajan and Dixit, 2010; Mahajan et al., 2012). This review has focused on the practical implications of using the tissue engineering techniques in medical and dental treatments and critically evaluates the ground reality behind the approach.
REGULATORY ISSUES
In the field of tissue engineering and regenerative medicine, government intervention begins before the start of research itself. There are certain regulatory and practical issues that need to be considered prior to developing the tissue engineering approach.
All regenerative products are regulated by FDA, a science based agency in the US Public Health Service (PHS) which have the legislative authority for the premarket approval and post market surveillance. The FDA consist of six centers that are staffed with individuals expert in the science and regulations, out of which three centers are concerned with medical products rest are involved in cosmetics and veterinary pharmaceuticals:
| • | Center for drug evaluation and research(CDER) |
| • | Center for biologics evaluation and research(CBER) |
| • | Center for devices and radiological health(CDRH) |
In addition to the centers there are:
| • | Offices for regulatory affairs (ORA) |
| • | Offices for orphan products(OOP) |
| • | Offices of combination product (OCP) (Smith and Hellman, 2003) |
FDA has classified medical product into-device drug or biologic or a combination product (i.e., a combination device/drug or device/biologic etc.) (Fig. 1) (Smith, 2006).
Drug: Drug is broadly defined as an article intended for use in the diagnosis, cure, mitigation, treatment or prevention of disease (FD and C Act §201(g)(1).
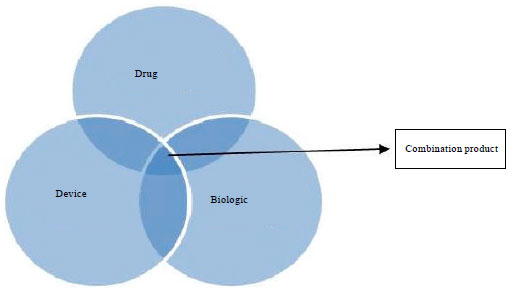 | |
| Fig. 1: | Classification of Biological tissue engineering products |
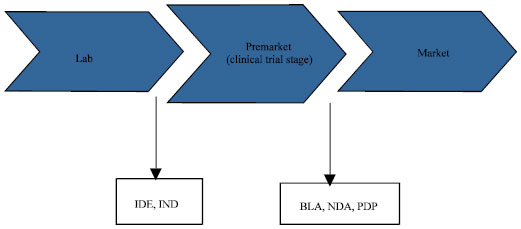 | |
| Fig. 2: | Various regulatory steps taken when a product is launched in the market |
Device: Device is an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent or other similar related article (FD and C Act, §201(h) (emphasis added).
Biologic: Biologic is defined as any virus, therapeutic serum, toxin, antitoxin, vaccine, blood, blood component or derivative, allergic or analogue product applicable for treatment and cure of disease (PHS Act, §351(a).
A combination product: Combination product poses a challenge to Food and Drug Administration (FDA), since they complicate the determination of the appropriate regulatory process. Disputes over the classification of a combination product between a sponsor and the FDA or between centers are submitted to FDA office for resolution (Lanza et al., 2006).
Implication of product classification: Certain medical products are simply what they are-like implant is obviously a device and aspirin is a drug but the classification of a new drug is essential as different products have different properties with minor variations in the measurement of safety and effectiveness. The product is subjected to various regulations prior to the launch in market (Fig. 2) (Smith, 2006).
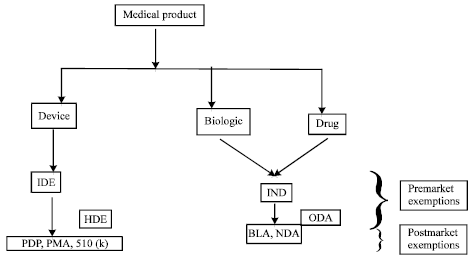 | |
| Fig. 3: | Various licence allocations and investigational device exemptions before it can be used on any human population, IDE: Investigational device exemption, HDE: Human device exemptions, PDP: Product development, PMA: Premarket application, IND: Investigational new drug, ODA: Orphan drug application, BLA: Biologic license application for biologic and drug, NDA: New drug application |
Similarly, any medical products, as per the classification (device, biologic and drug) need to undergo various screening steps. The products undergo different procedure of licence allocation and investigational device exemptions before it can be used in any human population (Smith, 2006) (Fig. 3).
PHASES OF CLINICAL TRIALS
Four phases of clinical trials and medicine development exist:
Phase 1: Initial safety trials on a new medicine. Pharmacokinetic trials are usually considered Phase I trials regardless of when they are conducted during a medicine's development.
Phase 2a: Pilot clinical trials to evaluate efficacy (and safety) in selected populations of patients with the disease or condition to be treated, diagnosed, or prevented.
Phase 2b: Well controlled trials to evaluate efficacy (and safety) in patients with the disease or condition to be treated, diagnosed, or prevented. These clinical trials usually represent the most rigorous demonstration of a medicine's efficacy.
Phase 3a: Trials conducted after efficacy of the medicine is demonstrated but prior to regulatory submission of a New Drug Application (NDA) or other dossier. These clinical trials are conducted in patient populations for which the medicine is eventually intended.
Phase 3b: Clinical trials conducted after regulatory submission of an NDA or other dossier but prior to the medicine's approval and launch. These trials may supplement earlier trials, complete earlier trials, or may be directed toward new types of trials.
Phase 4: This is the period between submission and approval of a regulatory dossier for marketing authorization.
Studies or trials conducted after a medicine is marketed to provide additional details about the medicine's efficacy or safety profile. Different formulations, dosages, durations of treatment, medicine interactions and other medicine comparisons may be evaluated. New age groups, races and other types of patients can be studied (Anonymous, 2009).
The key to success is selecting individual components of tissue engineering triad (cells, growth factors and scaffold) that has the best likelihood of obtaining regulatory approval (Liddell and Wallace, 2005).
Cells: Regulatory issues become more complex when moving from autogenous to allogenic cells. The FDA is mainly concerned with the characterization of the cells i.e., the product is being safe and consistent in its formulation.
Growth factor delivery system: Direct delivery is the easiest approach from a regulatory point of view. Gene enhanced cell delivery systems involves more regulatory hurdles.
Scaffold: Where possible matrix materials should be from non-mammalian sources (e.g., alginate). it is preferable to use materials which are already approved for clinical use. Otherwise the regulatory safety will make the phase 1 trial safety design more difficult.
PRACTICAL CONSIDERATIONS
Any new technology intended for widespread clinical use should be based on currently existing methodologies -allogenic cells must be expanded in culture, tested for genetic stability and infections prior to use.
In order to develop a cost effective autogenous tissue regenerative product, it is crucial to avoid technologies requiring ex vivo cell expansion.
Incubating innovation or cultivating corruption: Innovation has become a policy focus in many states as they compete to position themselves in the emerging knowledge economy. A substantial body of literature supports the growth of scientific misconduct in northeast Asia. The two distinct features of the innovation system are: liberal research regimes adopted by the government state and marked by freedom from government oversight and the illiberal laboratory cultures imported from German and marked by all powerful lab directors in East Asian companies (Lee et al., 2010).
Tissue market-profit based or evidence based???: FDA approves a drug only for a specified use and in a specified age group. It’s illegal to promote them for any other use. But physicians may prescribe approved drugs “off label”-i.e., without regard to specific use. Children as young as two years old are now being diagnosed with bipolar disorder and treated with a combination of powerful drugs, many of which were approved by FDA but none of which was approved below ten years of age. As drug companies don’t have direct access to human subjects, they go to medical schools for clinical trials, in part because the research is taken more seriously but it gives them access to highly influential faculty physicians. Furthermore they favor sponsored drugs- largely because they make their drug look better and safer than others by creating biases. It is not surprising that industry sponsored trials published in medical journals consistently-largely because the negative results are never published, positive results are repeatedly published in slightly different forms and positive spin is put on even negative results. A review of various bone grafts clinical trial found that 37 auto of 38 papers were published on the positive outcomes. It is not unusual for a published dapper to shift the focus from drug intended effect to secondary effects that are more favorable. Many drugs that are assumed to be effective are little better than placebos but there is no way out for this. One clue was provided by four researchers who reviewed every placebo controlled clinical trial study from FDA. They found that placebos were 80% as effective as the antidepressant drug itself .the difference between placebo and drug was so small that it was unlikely to be of any clinical significance but favorable results were published and unfavorable were buried (Lee et al., 2010).
Most of the sponsored drugs may be compared with other drugs administered at a dose so low that the sponsored drug look powerful or the drug of interest will be tested in the young population compared to the other drug in the older population so that the side effects are less likely to emerge. In recent years, drug companies have perfected a new and highly effective method to expand their markets. Instead of promoting drugs to treat disease, they have begun to promote disease to fit their product. People are told that they have medical conditions that require long term drug therapy sometimes called as drug mongering.
OBJECTIVES ONE SHOULD LOOK FOR BEFORE ADOPTING A NEW PRODUCT
Safety and quality-end users are primarily concerned with the safety and quality of product that it is not detrimental to the health.
Clinical efficacy or performance of claims: The product must ensure that the therapeutic potential of the new product is better or at least equivalent to the existing products.
Cost efficiency: Keep in mind that there should be at least enough sellers in the market for the similar products. Which ensures that the product definitely has therapeutic outcomes and the competition among the sellers will cut off the price to consumer’s level of satisfaction?
Social impact: Minority ethnic groups are sometimes reluctant to get benefits from tissue engineered products, furthermore most of the products are made by cost intensive technology that only the wealthy will be able to afford (Angell, 2009).
CONCLUSION
There is a very little systematic research and much of the research that existed and is scattered, inconsistent and ambiguous. The majority of us recognized that the amount of solid science and evidence on which we make our dissuasion is very less. So many reforms would be necessary to restore integrity to clinical research and medical practice. Many should involve congressional legislation and changes in the FDA including its drug approval process. But above all there is a need for the medical profession to wean of from industry money almost entirely.
There is again a need that every single clinician must try to get at least a basic background of the product including its approval and various steps that made it a tissue engineered product before its clinical application. Members of medical schools should not accept any payments from drug companies except research support, as industry-academic collaboration can make wonderful scientific contributions they should not spoil the essence of it by getting involved in the medical corruption. They are usually to carry out basic research and not the clinical trials.
REFERENCES
- Berthiaume, F., T.J. Maguire and L.Y. Martin, 2011. Tissue engineering and regenerative medicine: History, progress, and challenges. Ann. Review Chem. Biomol. Eng., 2: 403-430.
Direct Link - Galler, K.M. and R.N. D'Souza, 2011. Tissue engineering approaches for regenerative dentistry. Regen. Med., 6: 111-124.
PubMed - Lee, C.S. and A. Schrank, 2010. Incubating innovation or cultivating corruption?: The developmental state and life science in Asia. Oxford J., 88: 1231-1255.
CrossRefDirect Link - Liddell, K. and S. Wallace, 2005. Emerging regulatory issues for human stem cell medicine. Genomics Soc. Policy, 1: 54-73.
Direct Link - Mahajan, A., A. Bharadwaj and P. Mahajan, 2012. Comparison of periosteal pedicle graft and subepithelial connective tissue graft for the treatment of gingival recession defects. Aust. Dent. J., 57: 51-57.
CrossRefPubMedDirect Link - Mahajan, A., 2011. Periosteum- a highly under rated tool in dentistry. Int. j. dentist., 2012: 6-6.
CrossRef - Mahajan, A. and J. Dixit, 2010. Patient satisfaction with acellular dermal matrix graft in the treatment of multiple gingival recession defects-A clinical study. Webmedcentral, Clin. Trials, Dentistry, 30: 1-10.
Direct Link - Mahajan, A. 2009. Periosteal pedicle graft for the treatment of gingival recession defects: A novel technique Australian Dental J., 54: 250-254.
CrossRef - Moharamzadeh, K., I.M. Brook, R. Van Noort, A.M. Scutt and M.H. Thornhill, 2007. Tissue-engineered Oral Mucosa: A review of the scientific literature. J. Dent. Res., 86: 115-124.
CrossRefDirect Link - Smith, D. and K.B. Hellman, 2003. Regulatory oversight and product development: Charting the path. Tissue Eng., 9: 1057-1058.
CrossRef