
Wuyi Liu
Department of Science and Technology, Fuyang Normal University, No. 100 West Qing He Road, Fuyang, People`s Republic of China
LiveDNA: 86.3112
Biotechnology
Year: 2017 | Volume: 16 | Issue: 4-6 | Page No.: 145-154







ABSTRACT
Background and Objective: The regulatory gamma subunits of AMP-activated protein kinase (AMPK) are encoded by the 5'-AMP-activated protein kinase gamma subunit (PRKAG) genes. These genes are dominantly expressed in skeletal muscle and many reports of the animal counterparts suggest that these subunit genes play key roles in the regulation of energy metabolism in the skeletal muscle tissues. This study was designed to identify and analyze human and avian PRKAG genes in a genome scale. Materials and Methods: In the study, all the putative PRKAG gene sequences were used to construct ML (maximum likelihood) phylogenetic trees with package MEGA version 6.06 under JTT+I+G model. After phylogenetic analyses, the putative PRKAG gene were further subjected to the enrichment analyses of gene ontology (GO) and pathway annotations. In this study, there were totally 58 putative PRKAG genes and 31 unique PRKAG genes identified from the human and avian genomes. Results: Phylogenetic analyses indicated that among all the three sub-families of PRKAG genes (i.e. PRKAG1, PRKAG2 and PRKAG3), those protein sequences of PRKAG genes from four avian genomes formed monotonous phylogenetic clusters, whereas all the human protein sequences of PRKAG genes formed sole phylogenetic clusters. Furthermore, functional enrichment analyses of GO and pathway annotations revealed that PRKAG genes were functionally enriched in energy metabolism related signaling pathways and biological processes with significant p-values observed. Conclusion: The dataset and results of this study will facilitate further study on PRKAG genes in domestic animals.
PDF Abstract XML References Citation
Received: October 27, 2017;
Accepted: December 18, 2017;
Published: January 02, 2018
Copyright: © 2017. This is an open access article distributed under the terms of the creative commons attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
How to cite this article
Wuyi Liu, 2017. Genome-wide Identification and Analysis of Human and Avian 5'-AMP-Activated Protein Kinase Gamma Subunit Genes. Biotechnology, 16: 145-154.
DOI: 10.3923/biotech.2017.145.154
URL: https://scialert.net/abstract/?doi=biotech.2017.145.154
DOI: 10.3923/biotech.2017.145.154
URL: https://scialert.net/abstract/?doi=biotech.2017.145.154
INTRODUCTION
AMPK is a key sensor of cellular energy belonging to the serine-threonine kinase super-family presenting essentially all eukaryotic cells. AMPK appears as a heterotrimer composed of catalytic alpha (α) subunits and non-catalytic regulatory beta (β) and gamma (γ) subunits1-3. In the structural composition, alpha subunit has the kinase catalytic site (a conserved threonine residue in mammals) which is responsible for passing ATP phosphoric acid base group to the target proteins4. The alpha subunits comprise N and C functional regions that combine with the beta or gamma subunits at the C terminal region4. The gamma subunits comprise four duplicated tandem cystathionine-β-synthase (CBS) repeats or motifs responsible for the protein connection of AMPK with AMP, whereas the beta subunits serve as scaffolds and bracket alpha and gamma subunits with its anchoring domains5. On the other hand, many subtypes of AMPK are found to be encoded by distinct genes and animal genomes have genes encoding multiple isoforms of these subunits (α1, α2; β1, β2; γ1, γ2, γ3). The heterotrimers of AMPK are normally activated merely after phosphorylation of a conserved threonine residue within the activation loop of the alpha subunit kinase domain. Mammalian AMPK is activated through binding of 5'-AMP by the following three complementary effects6-10: The promotion of Thr172 phosphorylation by upstream kinases and a subsequent inhibition of Thr172 dephosphorylation by protein phosphatases and the final allosteric activation. Since ATP antagonizes these three complementary effects, AMPK acts as a sensor of cellular AMP:ATP and ADP:ATP ratios9. Thus, AMPK can sense small changes in AMP even in the presence of concentrations of ATP two to three orders of magnitude higher9,10. Furthermore, AMPK senses changes in AMP mainly through its direct binding to the gamma subunits. As AMPK gamma subunits contain four duplicated tandem CBS motifs, each tandem pair of CBS motifs often binds a regulatory adenosine-containing ligand like ATP and/or S-Adenosyl Methionine in the cleft between the paired motifs in the gamma subunits11. These four tandem motifs can assemble into a circular shape with one motif in each quadrant in AMPK gamma subunits, generating four potential nucleotide-binding sites11,12.
Since the activities of AMPK are important for cellular energy regulators and animal survival during periods of stress and metabolism and starvation, AMPK subunits, especially the gamma subunits, have been the focuses of many researchers dealing with both genetic and medical issues. The regulatory gamma subunits of AMPK are encoded by the 5'-AMP-activated protein kinase gamma subunit (PRKAG) genes. Those genes are dominantly expressed in skeletal muscle and many reports of the animal counterparts suggest that these subunit genes may play a key role in the regulation of energy metabolism in the skeletal muscle of pigs13-15 and chicken16-18. The first study of significant genetic mutation of PRKAG3 (AMPK γ3 subunit) genes was found and characterized in pigs with large effects on meat quality and processing yield13-15. Milan et al.13 identified and reported that a non-conservative substitution (R200Q) in the PRKAG3 gene locus significantly associated with excess glycogen content in the skeletal muscle of Hampshire pigs. Then, a second mutant allele (V199I) at the PRKAG3 gene locus was also identified and characterized, whereas its genetic effect on the meat quality of pork loin was reported in detail14,15. Other examples were found in chicken PRKAG genes, especially the PRKAG3 gene locus16-18. Zhao et al.16 found two SNPs (single nucleotide polymorphisms) in the 5'-end and 10 SNPs in exons 3, 4, 9 and 11 of chicken PRKAG3 gene locus, of which three caused amino acid substitutions, were identified in five chicken groups and/or breeds (i.e. Hubbard ISA White broiler, Leghorn layer, Tibet Chicken, Shouguang Chicken and Beijing Yellow Chicken)16. Proszkowiec-Weglarz et al.17 reported their work of molecular cloning, genomic organization and expression of three chicken 5'-AMP-activated protein kinase gamma subunit genes. Yin et al.18 studied the association of the genetic polymorphisms (SNPs) of PRKAG3 gene locus with the broiler carcass traits in 470 Chinese Daheng meat-type chickens and a nucleotide substitution (c. 3207 A>G) was detected in the eleventh exon of chicken PRKAG3 gene locus, which caused an amino acid change (p. 1069 T>A)18. They found that this SNP was significantly associated with the carcass traits live weight, carcass weight, breast muscle weight, leg muscle weight, abdominal fat weight and subcutaneous fat thickness18. Nevertheless, PRKAG genes have been a hotspot research topic and researchers carried out many studies on the genetic associations of PRKAG gene mutations with meat quality and other relevant traits in animals.
Given the genetic importance of PRKAG gene loci in animal sciences and the researchers' concern on PRKAG gene SNPS in animal genetics and nutrition and biotechnology, more genetic attention and studies should be conducted under the genomics era. However, no genome-wide research has been conducted to find how many PRKAG genes are encoded by the human and animal genomes, especially domestic animal genomes. This study was designed to carry out a genome-wide survey to identify and analyze the human and avian PRKAG genes from both the human and avian genomes.
MATERIALS AND METHODS
Data retrieve and sequence alignment: Firstly, the genome-wide method was used with three pair sequences of human PRKAG genes (PRKAG1, nucleotide sequence NM_001206709 with protein sequence NP_001193638; PRKAG2, nucleotide sequence NM_001040633 with protein sequence NP_001035723; PRKAG3, nucleotide sequence NM_017431 with protein sequence NP_059127) to perform TBLASTN searches against the five vertebrate genomes (i.e. Homo sapiens, Gallus gallus, Taeniopygia guttata, Meleagris gallopavo and Anas platyrhynchos) to retrieve all the putative PRKAG genes and proteins. Then, a number of closely related PRKAG genes have been isolated from these vertebrate species. ClustalX 2.019,20 was used to make gene sequence alignments and transformed sequences into an aligned coding fasta files of both nucleotide and protein sequences. Incomplete sequences and highly divergent regions or gaps would result in uncertain alignments and were excluded from the further analysis. Subsequently, more than 200 sequences were initially downloaded and a total of 58 gene sequences from 5 vertebrate species were identified and checked (the supplementary table). Finally, 31 unique PRKAG gene sequences were further checked and use and analyzed in detail. These final used data set included a total of 31 unique protein sequences from 5 different species (Table 1), including 18 sequences from human (Homo sapiens) genome, 6 sequences from chicken (Gallus gallus) genome, 4 sequences from Zebra Finch (Taeniopygia guttata) genome, 2 sequences from Turkey (Meleagris gallopavo) genome and 1 sequence from Mallard (Anas platyrhynchos) genome. Subsequent genome scale examination and phylogenetic analyses led us to successfully identify 31 unique PRKAG genes and define the orthologous PRKAG gene families with sufficient confidence.
Phylogenetic analyses of PRKAG genes: The initial 58 putative protein sequences and final 31 unique protein sequences of PRKAG gene loci were used to construct ML (maximum likelihood) phylogenetic trees with package MEGA version 6.0621 under a JTT+I+G model of evolution, respectively. Among those analyses, the bootstrap was set as 1,000 replicates in each ML tree and all the trees were further edited by package MEGA version 6.0621.
Functional enrichment analysis of gene ontology (GO) and pathway annotations: DAVID Functional Annotation Bioinformatics Microarray Analysis (URL: https://david. ncifcrf.gov/), is a web server for gene and/or protein functional annotation and functional set enrichment of gene with corrected p-values of enrichment and enrichment score based on statistical results of multiple methods22. All the putative PRKAG genes and/or protein sequences were input into the DAVID bioinformatics databases to retrieve potential functional information of PRKAG genes.
Based on the functional enrichment analyses of GO and pathway annotations and listed functional annotation results (including functional annotation chart, functional annotation clustering and functional annotation table) retrieved from the databases of DAVID Functional Annotation Bioinformatics Microarray Analysis, all the PRKAG genes can be functionally annotated and understood.
Statistical analysis: Statistical analyses for the enriched GO and pathway annotations were analyzed using the tool kits of DAVID bioinformatics databases that reported enrichment scores22, including a hyper-geometric p-value<0.05 and the Benjamini adjusted p-value<0.1, with respect to the ranked categories and clusters of GO and pathway annotations. Furthermore, some GO and pathway annotations were also listed and showed to be enriched with a hyper-geometric p-value around 0.05 and the Benjamini adjusted p-value around 0.1. Other statistical analyses were calculated by Microsoft Excel with the strict cutoff levels of p-value<0.05.
RESULTS AND DISCUSSION
Phylogenetic analyses of human and avian PRKAG genes: Determining the phylogenetic relationships of homologous and/or orthologous genes is an important step for elucidating the evolutionary and functional divergence of the gene family as well as that for individual genes. Meanwhile, with the genome sequence data for more and more animal species becoming available, it is now feasible to compare the whole gene family among different animal species at the genome-wide levels. Here, phylogenetic analyses of maximum likelihood tree (MLT) were used to identify putative homologous and/or orthologous relationships of the PRKAG gene family in 5 different species. The clusters of PRKAG gene family analyzed by phylogenetic trees were in agreement much with the traditional classification1,10, but more diversity was observed.
In the human and avian genomes, totally 58 pair putative PRKAG nucleotide and protein sequences were identified (supplementary Fig. 1 and supplementary Table 1) and finally 31 pair unique PRKAG nucleotide and protein sequences from these 5 vertebrate species were checked and used in phylogenetic analyses (Table 1).
 | |
| Supplementary Fig. 1: | Maximum likelihood phylogenetic tree of 58 human and avian protein sequences of PRKAG genes |
| Supplementary Table 1: | Fifty-eight nucleotide and protein sequences of PRKAG genes from 5 species (the gene information was identified from the NCBI genomic databases) |
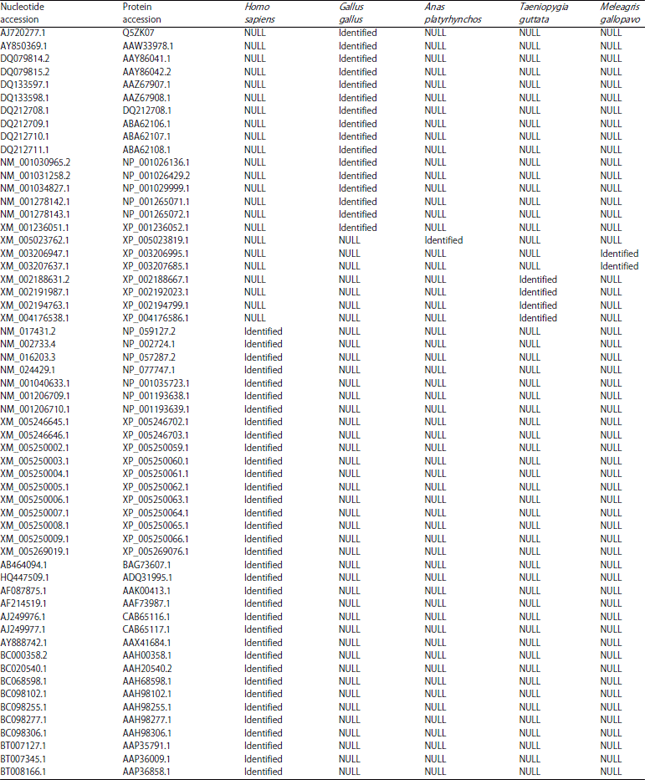 | |
Identified: This sequence ID was identified from the NCBI genomic databases used for corresponding species, NULL: This sequence ID was not used for corresponding species | |
| Table 1: | Thirty one unique nucleotide and protein sequences of PRKAG genes from 5 species |
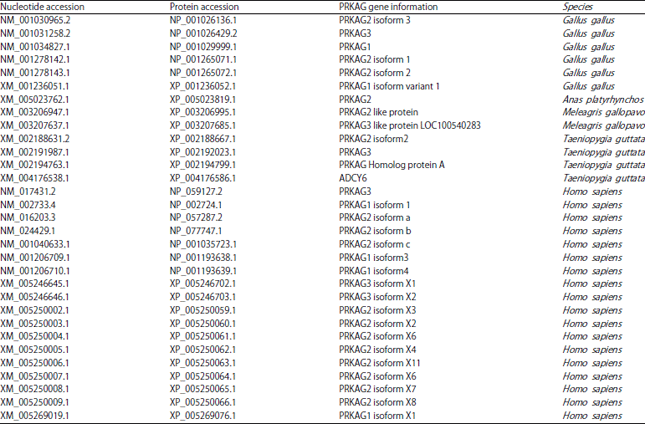 | |
| Gene information was identified from the NCBI genomic databases | |
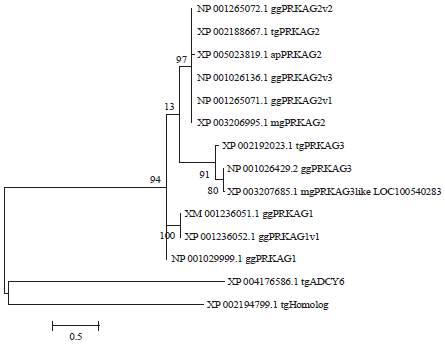
In the ML phylogenetic tree of 31 unique protein sequences of PRKAG genes, almost all the gene sequences were clustered according to their homologous sub-families and/or orthologous relationships regardless of their origination from different species (Fig. 1). Similar scenario was also observed in the ML phylogenetic tree of 58 putative protein sequences of PRKAG genes (supplementary Fig. 1). However, there were still subtle genetics and evolutionary differences between human and avian PRKAG genes, which could be inferred from the sub-clusters and evolutionary distances showed in these ML phylogenetic trees (Fig. 1; supplementary Fig. 1). From all the three sub-families of PRKAG genes (i.e. PRKAG1, PRKAG2 and PRKAG3), those protein sequences of PRKAG genes from four avian genomes formed monotonous phylogenetic clusters, whereas all the human protein sequences of PRKAG genes formed other sole phylogenetic clusters (Fig. 1; supplementary Fig. 1).
The avian protein sequences of PRKAG genes were further analyzed to explore their orthologous relationships and/or evolutionary distances (Fig. 2). It was revealed that sub-families PRKAG2 and PRKAG3 were closer than PRKAG1 deduced from the evolutionary distances in the ML phylogenetic tree (Fig. 2). Furthermore, among these avian PRKAG protein sequences, two protein sequences of predicted PRKAG genes (XP_002194799.1 and XP_004176586.1, PRKAG homolog proteins identified from Taeniopygia guttata) were discretely clustered (Fig. 2).
Functional enrichment analysis of human and avian PRKAG genes: All the 31 human and avian PRKAG gene sequences were input into DAVID Functional Annotation Bioinformatics Database22 to explore their functional annotations and enrichment and clustering groups. Using DAVID Functional Annotation Bioinformatics Tools, these 31 human and avian PRKAG gene sequences were functionally clustered and grouped and enriched according to the available GO annotations, genomic information from stored pathways and known gene-protein interactions (Table 2). All in all, functional annotation results of the present study agreed greatly with previous reports.
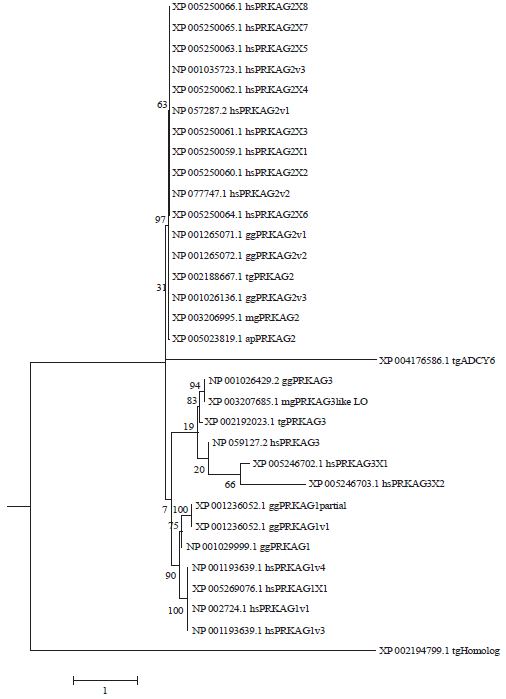 | |
| Fig. 1: | Maximum likelihood phylogenetic tree of 31 human and avian protein sequences of PRKAG genes |
| Table 2: | Functional enrichment analysis of the 31 unique PRKAG genes from 5 species mapped by the DAVID Functional Annotation Bioinformatics Tools |
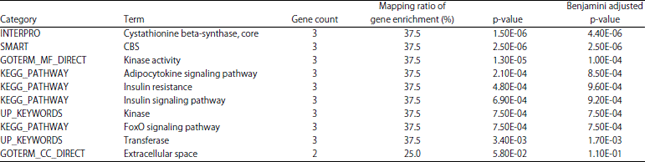 | |
| There is only one annotation cluster with the enrichment score as 3.91 provided by the DAVID Bioinformatics Databases | |
 | |
| Fig. 2: | Maximum likelihood phylogenetic tree of the avian protein sequences of PRKAG genes |
For instance, comparative transcriptome analysis of the Pacific White Shrimp (Litopenaeus vannamei) muscle reveals that most of the differentially expressed genes were involved in cell proliferation, growth and signaling, glucose homeostasis, energy and nutrients metabolism23. Functional enrichment analysis of these genes revealed AMPK signaling pathway and other 12 important pathways were identified as significantly enriched biological pathways23. These genes and pathways were involved in driving the Shrimp's feed efficiency23. Another research reported the identification of differentially expressed genes in chickens differing in muscle glycogen content and meat quality with Chicken Genome Arrays (20 K)24. They found several genes were suggested to be involved in cAMP-dependent signalling pathways and the regulation of glycogen storage in chicken muscle24. These differentially expressed genes, including PRKAB1 (AMPK β1 subunit), PRKAG2 (AMPK γ2 subunit) and UGDH (UDP-glucose dehydrogenase), may act as a glycogen sensor and compete for glycogen synthesis to impact on glycogen turnover in chickens muscle, through AMP-activated signalling pathways24.
In the present study, these 31 human and avian PRKAG gene sequences were significantly enriched in two GO annotations, i.e. (protein) kinase activity of molecular function (GOTERM_MF_DIRECT, GO:0016301, Benjamini adjusted p-value = 1.00E-04) and extracellular space of cellular component (GOTERM_CC_DIRECT, GO:0005615, Benjamini adjusted p-value = 1.10E-01), four KEGG pathways (Adipocytokine signaling pathway, Benjamini adjusted p-value = 8.50E-04; Insulin resistance, Benjamini adjusted p-value = 9.60E-04; Insulin signaling pathway, Benjamini adjusted p-value = 9.20E-04; FoxO signaling pathway, Benjamini adjusted p-value = 7.50E-04) and four other genomic database keywords (Table 2).
In the functional annotation clustering analysis, these 31 human and avian PRKAG gene sequences were only grouped into one functional enrichment cluster according to DAVID Functional Annotation Bioinformatics Tools (Table 2). It could be seen that these human and avian PRKAG genes mainly participate in energy metabolism related signaling pathways and biological processes (Table 2), such as adipocytokine signaling pathway, Insulin resistance and Insulin signaling pathway, FoxO signaling pathway, cystathionine beta-synthase, cystathionine-β-synthase (CBS), kinase, transferase and kinase activity. These resulted data of gene GO/pathway enrichment and functional annotation were in part agreement with the traditional functional opinions of AMPK kinases2-5,7-9. However, at present, there are few studies on the GO/pathway enrichment and functional annotation of PRKAG genes. As the coding genes of cellular energy sensing and regulating protein kinases, PRKAG genes have been intensely concerned and researched in animal skeletal muscle14-18,25-29 even since the report of a mutation in swine PRKAG3 associated with excess muscular glycogen content13. Recently, researchers are even more involved in functional analyses of gene mutation and enriched pathways of PRKAG genes with SNP and gene deletion/mutation reports25-29. For instance, a SNP in the 3'-untranslated region of PRKAG1 gene was identified as associate with serum ketone body and milk production of Holstein dairy cows25, whereas a SNP in the exon 11 of 5'-PRKAG3 gene was found to be associated with chicken meat water holding capacity26. In clinical investigations, mutation profiling identifies numerous rare drug targets and distinct mutation patterns in different clinical subtypes of breast cancers27 and PRKAG2 mutation was found as an easily missed cardiac clinical genotype of specific non-lysosomal glycogenosis28. In another research, physiological expression of AMPK-γ2RG (PRKAG2 with R531G) mutation causes Wolff-Parkinson-White (WPW) syndrome and induces kidney injury in mice, since cardiac-specific transgenic over-expression of human AMPK-γ2RG leads to constitutive AMPK activation and the WPW phenotype in mice29.
The present study discovered 58 putative PRKAG genes and 31 unique PRKAG genes and phylogenetic analyses verified all the three sub-families of PRKAG genes (i.e. PRKAG1, PRKAG2 and PRKAG3) in the human and avian genomes. The bird PRKAG gene sequences identified from four avian applied to resolve the critical issues of disease-induced loss that many researchers were not able to explore. These dataset and results formed in this study will also help and facilitate the further study on PRKAG genes in animals. However, the identified quantity of PRKAG genes and the researched amount and scope are insufficient in this study. Those are the limitations of the present study. More genes from recently sequenced genomes and more different species are need to be explored.
CONCLUSION
In the present study, there are totally 58 putative PRKAG genes and 31 unique PRKAG genes identified from the human and avian genomes. Phylogenetic analyses indicated that all the three sub-families of PRKAG genes (i.e. PRKAG1, PRKAG2 and PRKAG3), those protein sequences of PRKAG genes from four avian genomes formed monotonous phylogenetic clusters, whereas all the human protein sequences of PRKAG genes formed other sole phylogenetic clusters. The sub-families PRKAG2 and PRKAG3 were closer than PRKAG1 deduced from the evolutionary distances. Furthermore, functional enrichment analyses of GO and pathway annotations revealed that these PRKAG genes were functionally enriched in energy metabolism related signaling pathways and biological processes with significant p-values observed. In addition, these dataset and results will facilitate further study on PRKAG genes in animals. The study will provide useful information for future biological and medical studies on human and animal PRKAG genes.
SIGNIFICANCE STATEMENT
This study discovered 58 putative PRKAG genes and 31 unique PRKAG genes identified from the human and avian genomes. The study further discovers and explores the possible functional annotations of human and avian PRKAG genes that can be beneficial for relevant developmental and physiological and medical studies. This study will possibly help researchers to resolve the critical issues of disease-induced loss that many researchers were not able to explore. Meanwhile, these identified dataset and explored results formed in this study will also help and facilitate the further study on PRKAG genes in animals.
ACKNOWLEDGMENTS
Authors are grateful to the anonymous reviewers for their constructive comments and suggestions. It is jointly funded by National Natural Science Foundation of China (grant number No. 31301965) and Chinese research project of Anhui Provincial Educational Commission Natural Science Foundation (grant number No. KJ2016SD47).
REFERENCES
- Hardie, D.G., 2014. AMPK-sensing energy while talking to other signaling pathways. Cell Metab., 20: 939-952.
CrossRefPubMedDirect Link - Oakhill, J.S., J.W. Scott and B.E. Kemp, 2012. AMPK functions as an adenylate charge-regulated protein kinase. Trends Endocrinol. Metab., 23: 125-132.
CrossRefPubMedDirect Link - Carling, D., C. Thornton, A. Woods and M.J. Sanders, 2012. AMP-activated protein kinase: New regulation, new roles? Biochem. J., 445: 11-27.
CrossRefPubMedDirect Link - Hudson, E.R., D.A. Pan, J. James, J.M. Lucocq and S.A. Hawley et al., 2003. A novel domain in AMP-activated protein kinase causes glycogen storage bodies similar to those seen in hereditary cardiac arrhythmias. Curr. Biol., 13: 861-866.
CrossRefDirect Link - Hawley, S.A., M. Davison, A. Woods, S.P. Davies, R.K. Beri, D. Carling and D.G. Hardie, 1996. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem., 271: 27879-27887.
CrossRefPubMedDirect Link - Oakhill, J.S., R. Steel, Z.P. Chen, J.W. Scott, N. Ling, S. Tam and B.E. Kemp, 2011. AMPK is a direct adenylate Charge-regulated protein kinase. Science, 332: 1433-1435.
CrossRefDirect Link - Xiao, B., M.J. Sanders, E. Underwood, R. Heath and F.V. Mayer et al., 2011. Structure of mammalian AMPK and its regulation by ADP. Nature, 472: 230-233.
CrossRefPubMedDirect Link - Gowans, G.J., S.A. Hawley, F.A. Ross and D.G. Hardie, 2013. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab., 18: 556-566.
CrossRefPubMedDirect Link - Li, X., L. Wang, X.E. Zhou, J. Ke and P.W. de Waal et al., 2015. Structural basis of AMPK regulation by adenine nucleotides and glycogen. Cell Res., 25: 50-66.
CrossRefPubMedDirect Link - Scott, J.W., S.A. Hawley, K.A. Green, M. Anis and G. Stewart et al., 2004. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Invest., 113: 274-284.
CrossRefPubMedDirect Link - Xiao, B., R. Heath, P. Saiu, F.C. Leiper and P. Leone et al., 2007. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature, 449: 496-500.
CrossRefPubMedDirect Link - Milan, D., J.T. Jeon, C. Looft, V. Amarger and A. Robic et al., 2000. A mutation in PRKAG3 associated with excess glycogen content in pig skeletal muscle. Science, 288: 1248-1251.
CrossRefPubMedDirect Link - Lindahl, G., A.C. Enfalt, G. von Seth, A. Josell and I. Hedebro-Velander et al., 2004. A second mutant allele (V199I) at the PRKAG3 (RN) locus-I. Effect on technological meat quality of pork loin. Meat Sci., 66: 609-619.
CrossRefDirect Link - Lindahl G., A.C. Enfalt, G. von Seth, A. Joseli and I. Hedebro-Velander et al., 2004. A second mutant allele (V199I) at the PRKAG3 (RN) locus-II. Effect on colour characteristics of pork loin. Meat Sci., 66: 621-627.
CrossRefPubMedDirect Link - Zhao, C.J., C.F. Wang, X.M. Deng, Y. Gao and C.H. Wu, 2006. Identification of single-nucleotide polymorphisms in 5' end and exons of the PRKAG3 gene in Hubbard White broiler, Leghorn layer and three Chinese indigenous chicken breeds. J. Anim. Breed. Genet., 123: 349-352.
CrossRefPubMedDirect Link - Proszkowiec-Weglarz, M., M.P. Richards and J.P. McMurtry, 2006. Molecular cloning, genomic organization and expression of three chicken 5`-AMP-activated protein kinase gamma subunit genes. Poult. Sci., 85: 2031-2041.
CrossRefPubMedDirect Link - Yin, H.D., S.Y. Chen, Y. Wang, X.L. Zhao, Q. Zhu and Y.P. Liu, 2012. Association of protein kinase adenosine monophosphate-activated γ3-subunit (PRKAG3) gene polymorphisms with carcass traits in chinese meat-type chickens. J. Poult. Sci., 49: 254-259.
CrossRefDirect Link - Thompson, J.D., T.J. Gibson, F. Plewniak, F. Jeanmougin and D.G. Higgins, 1997. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res., 25: 4876-4882.
CrossRefPubMedDirect Link - Larkin, M.A., G. Blackshields, N.P. Brown, R. Chenna and P.A. McGettigan et al., 2007. Clustal W and clustal X version 2.0. Bioinformatics, 23: 2947-2948.
CrossRefPubMedDirect Link - Tamura, K., G. Stecher, D. Peterson, A. Filipski and S. Kumar, 2013. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol., 30: 2725-2729.
CrossRefPubMedDirect Link - Dennis, Jr., G., B.T. Sherman, D.A. Hosack, J. Yang, W. Gao, H.C. Lane and R.A. Lempicki, 2003. DAVID: Database for annotation, visualization and integrated discovery. Genome Biol., Vol. 4.
CrossRefDirect Link - Dai, P., S. Luan, X. Lu, K. Luo and J. Kong, 2017. Comparative transcriptome analysis of the pacific white shrimp (Litopenaeus vannamei) muscle reveals the molecular basis of residual feed intake. Sci. Rep., Vol. 7.
CrossRefDirect Link - Sibut, V., C. Hennequetantier, E.L. Bihanduval, S. Marthey, M.J. Duclos and C. Berri, 2011. Identification of differentially expressed genes in chickens differing in muscle glycogen content and meat quality. BMC Genomics, Vol. 12.
CrossRefDirect Link - Ahmad, M., Z. Amir, A. Hamid-Reza, A. Assad, V.H. Reza and E. Shahin, 2015. A SNP in the 3'-untranslated region of AMPKγ1 may associate with serum ketone body and milk production of holstein dairy cows. Gene, 574: 48-52.
CrossRefPubMedDirect Link - Yang, Y., D. Xiong, L. Yao and C. Zhao, 2016. An SNP in exon 11 of chicken 5'-amp-activated protein kinase gamma 3 subunit gene was associated with meat water holding capacity. Anim. Biotechnol., 27: 13-16.
CrossRefPubMedDirect Link - Santarpia, L., Y. Qi, K. Stemkehale, B. Wang, E.J. Young and D.J. Booser et al., 2012. Mutation profiling identifies numerous rare drug targets and distinct mutation patterns in different clinical subtypes of breast cancers. Breast Cancer Res. Treatment, 134: 333-343.
CrossRefPubMedDirect Link - Aggarwal, V., N. Dobrolet, S. Fishberger, J. Zablah, P. Jayakar and Z. Ammous, 2015. PRKAG2 mutation: An easily missed cardiac specific non-lysosomal glycogenosis. Ann. Pediatr. Cardiol., 8: 153-156.
CrossRefPubMedDirect Link - Yang, X., J. Mudgett, G. Bou-About, M.F. Champy and H. Jacobs et al., 2016. Physiological expression of AMPK&gamma2RG mutation causes wolff-parkinson-white syndrome and induces kidney injury in mice. J. Biol. Chem., 291: 23428-23439.
CrossRefPubMedDirect Link