
A.A.A. Hamid
Department of Industrial Biotechnology, Faculty of Biosciences and Bioengineering, University Technology Malaysia, 81310 Skudai, Johor, Malaysia
S. Hamdan
Department of Industrial Biotechnology, Faculty of Biosciences and Bioengineering, University Technology Malaysia, 81310 Skudai, Johor, Malaysia
Rolando V. Pakingking Jr.
Department of Aquaculture, Southeast Asian Fisheries Development Center, Tigbauan, Iloilo, 5021, Philippines
F. Huyop
Department of Industrial Biotechnology, Faculty of Biosciences and Bioengineering, University Technology Malaysia, 81310 Skudai, Johor, Malaysia
LiveDNA: 60.18518







ABSTRACT
Pseudomonas sp. strain S3 was isolated from Paddy (rice) field agricultural area. This organism, which can utilize a halogenated compound of D,L-2-Chloropropionic acid as sole carbon and energy source, catalyses the hydrolytic dehalogenation of both D- and L- isomers of 2-Chloropropionic acid. Identification of Pseudomonas sp. S3 is still ambiguous due to the lack of basic studies, especially their molecular genetic information. In this study, the amplified 16S rRNA gene sequence of Pseudomonas sp. S3 (Accession No. FJ968758) was compared to other nine selected gene sequences from the same group of Pseudomonas sp. and/or dehalogenase producing bacteria using in silico method. Their phylogenetic relationships were then determined. The results were analysed using MEGA4 software to ascertain its evolutionary distance by reconstructing a phylogenetic tree of these organisms. The evolutionary history and bootstrap consensus tree were inferred using the Neighbour-Joining method from 500 replicates. The tree is drawn to scale, with branch lengths (next to the branches) in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the p-distance method and were in the units of the number of base substitutions per site. Based on this analysis, Pseudomonas sp. S3 16S rRNA gene was closely related to the Pseudomonas chlororaphis with genetic distance 0.170 base substitutions per site. S3 gene was also compared among known dehalogenase producing bacteria 16S rRNA genes. Results suggested that S3 was closely related to the Pseudomonas sp. R1 with a genetic distance 0.040 base substitutions per site. From present study, evolutionary relationships of 16S rRNA gene of Pseudomonas sp. S3 were elegantly illustrated by phylograms, comparable to a pedigree showing which microorganisms are most closely related.
PDF Abstract XML References Citation
How to cite this article
A.A.A. Hamid, S. Hamdan, Rolando V. Pakingking Jr. and F. Huyop, 2010. Identification of Pseudomonas sp. Strain S3 Based on Small Subunit Ribosomal RNA Gene Sequences. Biotechnology, 9: 33-40.
DOI: 10.3923/biotech.2010.33.40
URL: https://scialert.net/abstract/?doi=biotech.2010.33.40
DOI: 10.3923/biotech.2010.33.40
URL: https://scialert.net/abstract/?doi=biotech.2010.33.40
INTRODUCTION
Each year, millions of tons of xenobiotic compounds are applied globally as herbicides in agricultural production area or in farmyards. As an outcome of this extensive environment input, natural water in rivers, lakes and aquifers has been contaminated with the trace amounts of herbicides compound. In Malaysia, organochlorine herbicides are widely used especially to clear weeds or unwanted crops. Environmental contamination of natural inland water have been a great concern, since, most of these herbicide compounds are very persistent, bioaccumulative and their toxicity can pose harmful effects to human and natural environment. Most of Southeast Asian countries like Malaysia, Thailand, Indonesia and Vietnam have banned the use of herbicide compounds since 1990s, but the residues are still detected in water and soil or sediments at the significant levels (Ibrahim et al., 2002).
Microbial catabolism of halogenated compound (Fig. 1) by dehalogenase producing bacteria has been well studied by Hardman (1991), Leisinger and Bader (1993), Janssen et al. (1994), Olaniran et al. (2001, 2004), Jing and Huyop (2007), Jing et al. (2008) and Ismail et al. (2008).
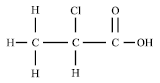 | |
| Fig. 1: | D, L-2-Chloropropionic acid |
A variety of halogenated compounds such as haloacids, which are produced by chemical industries, are degraded through dehalogenation by microbial dehalogenases that involve carbon-halogen bond cleavage (Copley, 1998).
Previous investigation suggested that two different kinds of dehalogenases were found in cells of Pseudomonas sp. strain S3 grown on D, L-2CP (Thasif et al., 2009). Both enzymes have great potential in biotransformation study. However, taxonomic identity and phylogeny of this unique halo-degrading bacterium has not been determined so far. Thus, we are interested in identifying the evolutionary relationship of this degradative bacteria among other Pseudomonas sp. group and/or dehalogenase producing bacteria in order to verify which bacteria was closely related and might be potentially degrade the same compound as Pseudomonas sp. strain S3. Using phylogenetic analysis, a complete 16S rRNA gene sequence of the Pseudomonas strain S3 was subjected to a specific computational tool for the gene sequences analysis and phylogenetic tree construction. Computational phylogenetics is the application of computational algorithms, methods and programs to phylogenetic analyses. MEGA4 molecular software was used in reconstructing a phylogram with the distance method approach to infer evolutionary distance of genes sequence. This software functionality has evolved to include the creation and exploration of sequence alignments, the estimation of sequence divergence, the reconstruction and visualization of phylogenetic trees and testing of molecular evolutionary hypothesis.
In this study, we have constructed two different phylograms which inferred the evolutionary relationship of Pseudomonas strain S3 to other Pseudomonas sp. and also their evolutionary distance among dehalogenase producing bacteria. The current goal is to assemble a phylogenetic tree representing a hypothesis about the evolutionary ancestry of a set of genes, species, or other taxa. The analysis will also able to predict the characteristics of an unknown microorganism based on their phylogenetic relationship.
MATERIALS AND METHODS
Characterization of Pseudomonas sp. S3: The 16S rRNA gene sequence of Pseudomonas sp. S3 was obtained by sequencing analysis. Chromosomal DNA of Pseudomonas sp. S3 was prepared and sent for sequencing (1st Base Laboratory Malaysia) using initial primers as described by Fulton and Cooper (2005). The 16S rRNA gene sequencing was subjected to MEGA4 computational tool in order to find regions of local similarity between selected sequences and also to generate a phylogenetic tree or phylogram which inferred evolutionary relationship. The 16S rRNA gene sequence of Pseudomonas sp. S3 was used to perform a BLAST search for homology study. From the selected of closely related sequences, a phylogram was established in Mega4 software in order to investigate the evolutionary pathway of Pseudomonas strain S3. In Mega4, homology analysis of selected sequences are aligned together by aligned Explorer/Clustal W. Phylogram was constructed using neighbor-joining method option (Tamura et al., 2007). Phylograms were rebuilt based on pairwise distance among sequence. Homology analysis of the 16S rRNA for S3 was also carried out among dehalogenase producing bacteria 16S rRNA gene and its phylogenetic tree was also constructed.
Collecting a set of homologous nucleotide sequences: The full 16S rRNA gene sequence from Pseudomonas sp. S3 was analyzed at http://www.ncbi.nlm.nih.gov/BLAST/, using BLASTn option. The BLASTn search will graphically displayed online on the Distribution of Blast Hits on the Query Sequence.
Building the phylogram of Pseudomonas strain S3 using BLAST web page: Phylogenetic tree of Pseudomonas sp. S3 and others evolutionary related bacteria were constructed using distance tree of results icon that available in same page of the list of homologous sequences (BLAST web page). The phylogenetic tree of Pseudomonas sp. S3 was presented and neighbour joining method was used to show relatedness and distance matrix of Pseudomonas sp. S3 in evolutionary pathway.
Reconstruction of phylogram using Mega4 software: Initially, a selected 16S rRNA gene sequences were converted into FASTA format and analyzed using alignment explorer/Clustal W in Mega4 (Fig. 2).
After completing sequence alignment by Clustal W, all output data were used to reconstruct phylogram. The evolutionary history was inferred using the neighbor joining method (Saitou and Nei, 1987). The Neighbor Joining (NJ) method constructs the tree by sequentially finding pairs of neighbors, which are the pairs of Operational Taxonomic Units (OTUs) connected by a single interior node. The clustering method used by this algorithm is quite different and does not attempt to cluster the most closely related OTUs, but rather minimizes the length of all internal branches and thus, the length of the entire tree.
 | |
| Fig. 2: | Multiple sequence alignment analysis by Clustal W |
The bootstrap consensus tree inferred from 500 replicates is taken to represent the evolutionary history of the taxa analyzed. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) are shown next to the branches (Felsenstein, 1985). The tree is drawn to scale, with branch lengths (next to the branches) in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the p-distance method (Tamura et al., 2004) and are in the units of the number of base substitutions per site. All positions containing gaps and missing data were eliminated from the dataset (Complete deletion option).
RESULTS
Identification of Pseudomonas strain S3: A bacterial species designated as S3 was isolated from paddy (rice) field agricultural area. The bacteria were grown aerobically at 30°C in 100 mL of a liquid minimal medium containing D, L-2-Chloropropionic acid as sole source of carbon. The 16S rRNA gene analysis was sequenced using FD1 and rP1 primers (Fulton and Cooper, 2005). The sequence comprises of 1469 nucleotides lacking the very proximal 5’ and terminal 3’ regions corresponding to the universal primers used and was submitted to the GenBank with accession number of FJ968758.
The S3 16S rRNA gene sequence was subjected to BLAST search (BLASTn). The sequence was compared with other sequences that contained in library database. This technique provides an algorithm for comparing primary biological sequence information. From BLASTn results, the most similar sequence was matched to the Pseudomonas sp. R1 (accession No. EU272817) with 98% sequence identity (Fig. 3). Hence, the bacteria was then designated as Pseudomonas sp. S3.
A phylogenetic tree of Pseudomonas sp. S3 and related species was also established using BLAST output homepage (Distance tree). From the phylogram, Pseudomonas sp. S3 located in the clade within others Pseudomonas sp. (Fig. 4). Strain S3 was diverged from a same node with Pseudomonas sp. strain R1. Strains S3 and R1 are sisters group which had minimum genetic distance.
Evolutionary relationship of Pseudomonas sp. S3 among Pseudomonas sp.: In this study, ten different Pseudomonas sp. from each major group of Pseudomonas were selected as operational taxonomic units (OTUs) in order to investigate the evolutionary relationship of Pseudomonas sp. S3 among other Pseudomonas sp. from various major groups. There are 14, 678 base nucleotides from various 16S rRNA gene of Pseudomonas sp. were analyzed and multiple alignment were constructed using Clustal W. All results were based on the pairwise analysis of 10 sequences. The longest genetic distance was between sequence of Pseudomonas sp. S3 and Pseudomonas luteola (0.199) while the shortest pairwise distance was between Pseudomonas sp. S3 and Pseudomonas chororaphis (0.170) as in Fig. 5. The data was generated based on the proportion of different homologous sites known as observed distance or p-distance and it is expressed as the number of nucleotide differences site. All positions containing gaps and missing data were eliminated from the dataset (Complete deletion option). There were a total of 1309 positions in the final dataset.
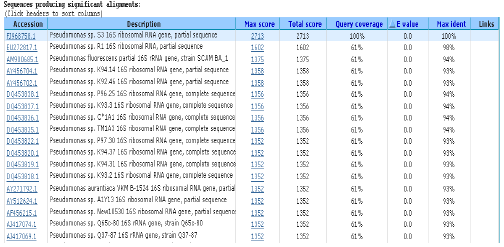 | |
| Fig. 3: | The 16S rRNA gene identity using BLAST output obtained from NCBI database |
 | |
| Fig. 4: | Phylogenetic tree of Pseudomonas sp. S3 among related species from reconstructed of BLAST results |
Data set illustrated in Fig. 5. was then converted into a proper phylogenetic analysis (Fig. 6) suggesting Pseudomonas sp. S3 was not located within the clade of other Pseudomonas species. Operational Taxonomic Units (OTUs) was selected from different major groups of Pseudomonas sp.
From all selected species, Pseudomonas chlororaphis from P.chlororaphis group was closely related to the Pseudomonas sp. S3 (Fig. 6). Pseudomonas chlororaphis is known as a biocontrol agent against certain fungal plant pathogens via production of phenazine type antibiotics (Chin-A-Woeng et al., 2000). Other Pseudomonas sp. that closely related to the S3 was Pseudomonas syringae strain BC2366. Pseudomonas syringae is a rod shaped Gram-negative bacterium with polar flagella. It is also a member of the Pseudomonas genus and placed in the Pseudomonas syringae group (Anzai et al., 2000).
 | |
| Fig. 5: | The number of base substitutions per site from analysis between sequences. All results are based on the pairwise analysis of 10 sequences. Analyses were conducted using the p-distance method in MEGA4. All positions containing gaps and missing data were eliminated from the dataset (Complete deletion option). There were a total of 1309 positions in the final dataset |
Pseudomonas syringae is a plant pathogen which can infect a wide range of plant species. Pseudomonas sp. S3 has distance relationship with Pseudomonas luteola. Pseudomonas luteola is a Gram-negative, rod-shaped, motile bacterium that can cause peritonitis, cellulitis and bacteremia (Kodama et al., 1985). It has also been shown to reduce and hence decolorize azo dyes (Hu, 2001). Based on 16S rRNA analysis, Pseudomonas luteola has been placed in the Pseudomonas stutzeri group.
Evolutionary relationship of Pseudomonas sp. among dehalogenase producing bacteria: In this study, ten dehalogenase producing bacteria were selected. The dehalogenase producing bacteria were comprised of Pseudomonas strain R1 (EU272817), Pseudomonas strain S3 (FJ968758), Pseudomonas corrugata strain SB4 (AY050495), Methylobacterium extorquen strain DM4 (AF227128), Comamonas sp. strain CY01 (EU515237), Stenotrophomonas maltophilia strain SB5 (AY050496), Methylobacterium strain HN2006B (AM231910), Serratia marcescens strain HL1 (EU371058), Rhodococcus strain HN2006A (AM231909) and Dehalococcoide strain BAV1 (AY165308).
There was limited number of data of 16S rRNA gene sequence published in the database for dehalogensase producing bacteria. All the gene sequences were obtained from NCBI data base. There were 13, 318 base nucleotides of 16S rRNA gene from ten different dehalogenase producing species were analyzed using multiple sequences alignment constructed by Clustal W. The number of base substitutions per site was shown in Fig. 7. One of the closest species was between Methylobacterium HN2006b and Methylobacterium extorquens apart from S3 and R1. All results were based on the pairwise analysis of 10 sequences conducted using the p- distance method with 624 positions in the final dataset.
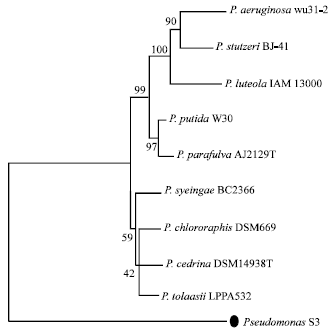 | |
| Fig. 6: | Phylogenetic tree showing evolutionary relationships of Pseudomonas sp. S3 among Pseudomonas species that was determined by the analysis of 16S rRNA gene sequences. The scale bar represents 0.005 substitutions per site. Bootstrap values above 40% are shown at the nodes (based on 500 resamplings) |
Figure 8 showed all the dehalogenase producing bacteria in the phylogenetic tree presented as Operational Taxonomic Units (OTUs) or external nodes. There are eight Hypothetical Taxonomic Units (HTUs) or internal nodes that illustrated in the bootstrap consensus phylogram. Pseudomonas sp. S3 and Pseudomonas sp. R1 are sisters group because they sharing a common node or ancestor. Pseudomonas sp. S3 was clustered with a good bootstrap support (100%) within a clade consisting Pseudomonas sp. R1 and Pseudomonas corrugata strains SB4. The results also suggested that Pseudomonas sp. R1 was the most closely related to the Pseudomonas sp. S3 with a genetic distance of 0.040 substitutions per site. Pseudomonas sp. R1 is a soil pseudomonad that able to degrade monochloroacetic acid (MCA) (Ismail et al., 2008). Other related species that located in the same clade as Pseudomonas sp. S3 was Pseudomonas corrugata strain SB4. This species was diverged from Pseudomonas sp. S3 at 0.082 base substitutions per site. Pseudomonas corrugata strain SB4 was isolated from soil contain a mixture of aniline and 4-chloroaniline (4CA) as principal carbon sources (Radianingtyas et al., 2003).
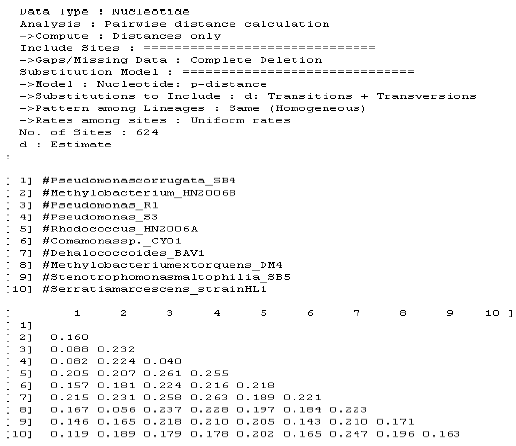 | |
| Fig. 7: | The number of base substitutions per site from analysis between sequences is shown. All results are based on the pairwise analysis of 10 sequences |
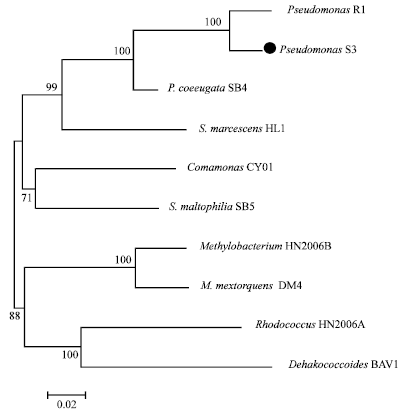 | |
| Fig. 8: | The phylogenetic tree showing evolutionary relationships of Pseudomonas sp. S3 among dehalogenase producing bacteria that was determined by the analysis of 16S rRNA gene sequences. The scale bar represents 0.02 substitutions per site. Bootstrap values above 70% at the nodes (based on 500 resamplings) |
DISCUSSION
Pseudomonas sp. have ability to use many organic compounds as carbon and energy sources. On the other hand, Pseudomonas generally lacks the hydrolytic enzymes necessarily to break down polymer into their component monomers. These nutritionally versatile pseudomonads contain numerous inducible operons because the catabolism of unusual organic substrates often requires the activity of several different enzymes. Pseudomonads are ecologically importants organisms in soil and water and are probably responsible for degradation of many soluble compounds derived from breakdown of plant and animal materials. They are also capable of breaking down many xenobiotic compounds, such as pesticides and other toxic chemicals and thus, important agent of bioremediation in the environment.
S3 was originally isolated from a paddy rice field and capable of degrading the D, L-2-Chloropropionic acid. The bacterial species was identified as gram negative rods in chains (Thasif et al., 2009). The identity of the bacteria was studied using phylogram and evolved from the same ancestor with others Pseudomonas sp. Using molecular analysis of phylogenetic software, strain S3 was closely related to the genus Pseudomonas chlororaphis group. This is possibly due to this strain S3 have the same characteristics to the species that belonged into this group which can degrade pollutants.
Strain S3 was also closely related to the Pseudomonas sp. strain R1. Both strains produced dehalogenases. Pseudomonas sp. S3 and R1 were classified as gram negative bacteria, motile and also ubiquitous in soil and water. Both bacteria produced dehalogenase enzyme and could degrade halogenated compound as a carbon source. Even though both organisms were closed relationship, but their enzyme characteristics were not identical. Pseudomonas sp. S3 for example produced both types of dehalogenase specific to D- and L- isomer whereas, Pseudomonas sp. R1 only produced dehalogenase that is non-stereospecific (Ismail et al., 2008).
Pseudomonas sp. S3 has distant relationship to Dehalococcoide sp. BAV1 even though they were classified as Dehalogenase producing bacteria. Their features indicated that both bacteria were not similar in morphology as expected. Dehalococcoides was related to the green non-sulfur bacteria (Chloroflexus group). Dehalogenase from Dehalococcoides preferred to de-chlorinated ethenes rather than chlorinated aliphatic acid compound (He et al., 2003).
In conclusion, this study provides the identity of D, L-2-Chloropropionic acid degrading bacteria using phylogenetic study. The evolutionary relationship of Pseudomonas sp. S3 has been depicted from computational program based on the molecular phylogenetic. The current research is very useful in identifying the genus of the isolated bacterial species. From the results, it was also possible to predict their general properties for further characterization.
ACKNOWLEDGMENT
This study was partly supported by Fundamental Research Grant Schemes 78307 from Ministry of Higher Education (MOHE) Malaysia.
REFERENCES
- Anzai, Y., H. Kim, J.Y. Park, H. Wakabayashi and H. Oyaizu, 2000. Phylogenetic affiliation of the pseudomonads based on 16S rRNA sequence. Int. J. Syst. Evol. Microbiol., 50: 1563-1589.
PubMed - Chin-A-Woeng, T.F.C., G.V. Bloemberg, I.H.M. Mulders, L.C. Dekkers and J.J. Ben, 2000. Root colonization by phenazine-1-carboxamide-producing bacterium Pseudomonas chlororaphis PCL1391 is essential for biocontrol of tomato foot and root rot. Mol. Plant Microbe Interact., 13: 1340-1345.
CrossRefDirect Link - Felsenstein, J., 1985. Confidence limits on phylogenies: An approach using the bootstrap. Evolution, 39: 783-791.
CrossRefDirect Link - Fulton, C.K. and R.A. Cooper, 2005. Catabolism of sulfamate by Mycobacterium sp. CF1. Environ. Microbiol., 7: 378-381.
Direct Link - Hardman, D.J., 1991. Biotransformation of halogenated compounds. Crit. Rev. Biotechnol., 11: 1-40.
CrossRef - He, J., K.M. Ritalahti, M.R. Aiello and F.E. Loffler, 2003. Complete detoxification of vinyl chloride (VC) by an anaerobic enrichment culture and identification of the reductively dechlorinating population as a Dehalococcoides species. Applied Environ. Microbiol., 69: 996-1003.
PubMed - Hu, T.L., 2001. Kinetics of azoreductase and assessment of toxicity of metabolic products from azo dyes by Pseudomonas luteola. Water Sci. Technol., 43: 261-269.
PubMed - Ibrahim, M.S., G. Jacinto, D. Connell and L.K. Leong, 2002. Regionally based assessment of persistent toxic substances region. VIII-Southeast Asia and South Pacific. Proceedings of Workshop on Environmental Monitoring of Persistent Organic Pollutants (POPs) in East Asian Countries, December 2-4, 2002, Tokyo, pp: 146-154.
- Ismail, S.N., A.M. Taha, N.H. Jing, R.A. Wahab, A.A. Hamid, R.V. Pakingking Jr. and F. Huyop, 2008. Biodegradation of monochloroacetic acid by a presumptive Pseudomonas sp. strain R1 bacterium isolated from Malaysian paddy (rice) field. Biotechnology, 7: 481-486.
CrossRefDirect Link - Janssen, D.B., F. Pries and J. van der Ploeg, 1994. Genetics and biochemistry of dehalogenating enzymes. Ann. Rev. Microbiol., 48: 163-191.
PubMed - Jing, N.H., A.M. Taha, R.V. Pakingking, Jr., R.A.B. Wahab and F. Huyop, 2008. Dehalogenase from Methylobacterium sp. HJ1 induced by the herbicide 2,2-dichloropropionate (Dalapon). Afr. J. Microbiol. Res., 2: 32-36.
Direct Link - Jing, N.H. and F. Huyop, 2007. Dehalogenation of chlorinated aliphatic acid by Rhodococcus sp. Asia Pac. J. Mol. Biol. Biotechnol., 15: 147-151.
Direct Link - Kodama, K., N. Kimura and K. Komagata, 1985. Two new species of Pseudomonas: P. oryzihabitans isolated from rice paddy and clinical specimens and P. luteola isolated from clinical specimens. Int. J. Syst. Bacteriol., 35: 467-474.
CrossRef - Leisinger, T. and R. Bader, 1993. Microbial dehalogenation of synthetic organohalogen compounds: Hydrolytic dehalogenases. Chimia, 47: 116-121.
Direct Link - Olaniran, A.O., G.O. Babalola and A.I. Okoh, 2001. Aerobic dehalogenation potentials of four bacterial species isolated from soil and sewage sludge. Chemosphere, 45: 45-50.
CrossRef - Olaniran, A.O., D. Pillay and B. Pillay, 2004. Haloalkane and haloacid dehalogenases from aerobic bacterial isolates indigenous to contaminated sites in Africa demonstrate diverse substrate specificities. Chemosphere, 55: 27-33.
CrossRef - Radianingtyas, H., G.K. Robinson and A.T. Bull, 2003. Characterization of a soil-derived bacterial consortium degrading 4-chloroaniline. Microbiology, 149: 3279-3287.
CrossRef - Saitou, N. and M. Nei, 1987. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol., 4: 406-425.
CrossRefPubMedDirect Link - Tamura, K., M. Nei and S. Kumar, 2004. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. U.S.A., 101: 11030-11035.
CrossRefDirect Link - Tamura, K., J. Dudley, M. Nei and S. Kumar, 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol., 24: 1596-1599.
CrossRefPubMedDirect Link - Thasif, S., S. Hamdan and F. Huyop, 2009. Degradation of D,L-2-chloropropionic acid by bacterial dehalogenases that shows stereospecificity and its partial enzymatic characteristics. Biotechnology, 8: 264-269.
CrossRefDirect Link