
S. Sur
NBU Bioinformatics Faculty, University of North Bengal, Siliguri 734013, India
G. Sen
NBU Bioinformatics Faculty, University of North Bengal, Siliguri 734013, India
S. Thakur
NBU Bioinformatics Faculty, University of North Bengal, Siliguri 734013, India
A.K. Bothra
Department of Chemistry, Raiganj College, Raiganj 733134, India
A. Sen
NBU Bioinformatics Faculty, University of North Bengal, Siliguri 734013, India
Biotechnology
Year: 2009 | Volume: 8 | Issue: 4 | Page No.: 434-441







ABSTRACT
Availability of the sequences of latest strains of H1N1 virus and their comparison with other viral strains may provide significant clues to the nature of H1N1. The objective of the study was to look into the characteristics of genes and proteins of the swine flu and related viruses to understand their lifestyle and evolutionary relationship. Sequences of genome segments were analysed using ACUA, Codon W and DAMBE. Evolutionary relationships were determined via condensed matrix method. CAI values were quite high in the studied viruses and pI values of proteins showed a bi-modal distribution. All HIN1 strains as well as Influenza C, H3N2 and H2N2 had pI in the range greater than 8.1 with H1N1 CAL07/2009 having pI value of 8.87. Positive correlations of GC3 and GC content with CAI values were noticed. Hydropathy and aromaticity levels increased with the decrease of GC3. Phylogram revealed a rooted tree, which shows two major clades, Clade A and Clade B with subclades. Majority of H1N1 lie together in the same clade with the exception of H1N1 CAL04/2009 that lies in a different clade altogether along with H1N1 Puerto-Rico. Mutational bias is the main factor driving codon usage variation. High expression of pathogenicity related genes confirm its role as pathogen. Most of the H1N1 basic proteomes are influenced by mutational pressure. Genes associated with the hydrophilic proteins are favoured by translationally optimal codons. Phylogenetic analysis portrays the role played by reassortment in controlling the evolution of the studied strains.
PDF Abstract XML References Citation
How to cite this article
S. Sur, G. Sen, S. Thakur, A.K. Bothra and A. Sen, 2009. In Silico Analysis of Evolution in Swine Flu Viral Genomes Through Re-assortment by Promulgation and Mutation. Biotechnology, 8: 434-441.
DOI: 10.3923/biotech.2009.434.441
URL: https://scialert.net/abstract/?doi=biotech.2009.434.441
DOI: 10.3923/biotech.2009.434.441
URL: https://scialert.net/abstract/?doi=biotech.2009.434.441
INTRODUCTION
Influenza or flu is caused by a group of viruses called influenza viruses. These categories of A, B and C viruses are the common pathological agents (Suzuki and Nei, 2002). Genomes of these viruses are segmented, single stranded and have (-) RNA. They are associated with epidemics and pandemics in mammals and birds. Wild waterfowl and other aquatic birds are the natural reservoirs of influenza viruses (Holmes et al., 2005). Influenza epidemics are accountable for causing 10,000-15,000 deaths in humans every year (Holmes et al., 2005).
Swine influenza also called swine flu is caused by a strain of influenza virus named H1N1 that usually infect pigs. Pandemic influenza has created havoc in 1918 (HIN1), 1957 (H2N2) and 1962 (H3N2) resulting in numerous deaths worldwide (Cox and Subbarao, 2000; Webby and Webster, 2003), while H5N1 assumed epidemic proportions in Asia in the years 2003-2005 (Holmes et al., 2005). Very recently, outbreaks of swine flu have sent shock waves in Mexico and United States, with the World Health Organization (WHO) issuing warning for possibility of worldwide pandemic. Although, the origin of this strain is still unknown, some reports point out that it has not been found in pigs. WHO reported (www.who.int/mediacentre/news/statements/2009/H1N1-20090427) that the mutated form of the virus might have been transmitted between humans and causes symptoms of influenza, such as runny nose, fever, coughing and headache etc. The H1N1 form of swine flu is reported to be a form of the causative agent that caused the pandemic in humans in 1918-1919 (Taubenberger and Morens, 2006). The descendants of the 1918 H1N1 virus have persisted among humans as well as pigs throughout the 20th century, with some seasonal bouts of influenza (Taubenberger and Morens, 2006). New variants of influenza viruses arising out of reassortment of the segmented RNA genome pose severe threat to public health (Gog et al., 2007). The 2009 swine flu strain of influenza is reported (www.inspection.gc.ca/english/ corpaffr/newcom/2009) to be a reassortment of four strains that includes influenza A virus subtype H1N1, one endemic in humans, one in birds and two in swine.
The availability of the sequences of the segments of some of the latest strains of H1N1 virus has given an opportunity to look into the pattern of codon usage, gene expression levels, determine protein isoelectric points, aromaticity and hydropathicity indicates the solubility of the proteins: positive GRAVY (hydrophobic), negative GRAVY (hydrophilic) of the amino-acids in addition to studying their molecular phylogeny.
Synonymous codons are unequally used between genomes. Compositional bias, translational selection and mutational pressure account for codon usage variation amongst organisms (Sur et al., 2008). Highly expressed genes have tendency for codons with high concentration of related tRNA molecules, whereas low expressed ones have consistent codon usage (Zhou et al., 2005). It has been reported that mutational pressure plays a significant role in influencing codon patterns in human viruses (Zhou et al., 2005). Estimation of the CAI (codon adaptation index) values (Sen et al., 2008) indicates the nature of gene expression levels of the respective genes. The physical properties of the proteins are fundamental in the normal functioning of an organism (Knight et al., 2004). Properties such as isoelectric point, hydropathicity and aromaticity play a role in protein functioning. Environment and GC content is known to play a crucial part in influencing amino-acid usage in organisms (Tekaia and Yeramian, 2006). The correlation of GC3 and GC content of the organisms with isoelectric point and amino acid frequencies of the proteins is expected to throw light into the molecular nature of swine flu viruses. The results obtained for the latest strains will be compared with that of the older strains of H1N1 and other flu viruses like H5N1, H2N2, H9N2, H3N2, influenza B and influenza C virus.
On the other hand phylogeny study of swine flu and other related viruses will shed some light on their evolutionary relationship. An important method of phylogeny developed by a group of laboratories including ours use nucleotide triplet based condensed matrix method (Mondol et al., 2008). Phylogenetic studies using sequence alignment and structures are insufficient in portraying the evolution of genes given that sequence comparison becomes unreliable at identity levels lower than 25% (Mondol et al., 2008). It also turns out to be tough to distinguish among properly aligned homologs and discrete sequences. Structure based methodologies are also insufficient given that number of structures are scarce to represent any significant conclusion. The condensed matrix method that relies on nucleotide triplet based phylogeny, is free from the aforesaid limitations as it takes into account, full length of the genes for creating phylograms (Mondol et al., 2008).
In a nutshell the aim of the present study is to look into the important characteristics of the genes and proteins of the swine flu and related viruses to infer upon their lifestyle and evolutionary relationships.
MATERIALS AND METHODS
The research work was started in the spring of 2009. It was virtually done in two laboratories. The software was developed in Department of Chemistry, Raiganj College. All bioinformatics analysis were performed at NBU Bioinformatics Facility, NBU while interpretation of results and paper writing were done in both the laboratories.
Sequences of the genome segments of Influenza A viruses [A/California/04/2009(H1N1); A/California/05/2009 (H1N1); A/California/07/2009(H1N1); A/Texas/04/2009 (H1N1) and A/Texas/05/2009(H1N1)] were obtained from the NCBI website (http://www.ncbi.nlm.nih.gov) and that of other Influenza A viruses like [A/Goose/Guangdong/ 1/96(H5N1); A/Korea/426/68 (H2N2); A/Hong Kong/1073/99(H9N2); A/New York/392/ 2004(H3N2); A/Puerto Rico/8/34(H1N1)] and Influenza B virus and Influenza C virus were obtained from the IMG database (http://img.jgi.doe.gov/cgi-bin/pub/main.cgi) (Markowitz et al., 2006).
The ACUA (Umashankar et al., 2007) was utilized to compute GC content, GC3 composition (amount of G or C codons in the third position), Nc (Effective number of codons) and CAI (codon adaptation index) values. The Nc measures codon bias whose value ranges from 20 to 61 (Sur et al., 2009). The CAI computes the relative adaptation of codon usage of genes towards codon usage of highly expressed genes (Wu et al., 2005). The CAI values vary from 0 to 1 with higher values signifying that, gene of concern has a codon usage pattern analogous to highly expressed genes. Certain viruses like bacteriophages have their own tRNA and it is inferred that though phages use most of the cells translational machinery and complement it with their own genetic information to attain higher fitness (Bailly-Bechet et al., 2007). However, in swine flu viral genomes there is no gene coding for any tRNA assuming that the swine flu viruses entirely depend upon host cells translational machinery. Therefore, we have used codon usage table of Homo sapiens as a reference for determining CAI values. Hydropathicity (GRAVY score) and aromaticity of the genes were determined using Codon W (http://mobyle.pasteur.fr/cgi-bin/MobylePortal/portal.py?form=codonw) (Peden, 1999). Hydropathicity or GRAVY score is calculated as the arithmetic mean of the sum of hydropathic indices of each amino acid, whereas aromaticity determines amino acid usage, provided inequality in amino acid composition includes application for evaluating codon usage (Lobry and Gautier, 1994). Distribution of isoelectric points (pI) in a proteome is one of the most important aspects of proteins (Kiraga et al., 2007). Protein isoelectric points were calculated using DAMBE (http://dambe.bio.uottawa.ca). Correspondence analysis was computed for codon usage on codon count using Codon W
(http://mobyle.pasteur.fr/cgi-bin/MobylePortal/portal.py?form=codonw) (Peden, 1999). Major trends in codon usage variation among the genes within the genomes are estimated with this analysis.
Determination of frequency of triplets of nucleic acid bases: Our own program written in Turbo C++ was used to count all the possible triplets of the nucleotide sequences of the whole genomes from the studied viruses. The matrices were formed using all the triplets. Introduction of a 4x4x4 cubic matrix having 64 possible entries resolves the frequency of incidence of all the possible 64 triplets in a DNA sequence. Here, it is possible to obtain three groups of 4x4 matrices {M1, M2, M3, M4}, {M5, M6, M7, M8}, {M9, M10, M11, M12} each one having every entry of cubic matrix (Randic et al., 2001). Usually, the group of 4x4 matrices {M1, M2, M3, M4} are taken as representative of the cubic matrix. Our technique gives equal weight to all positions since it considers nucleotide triplets, not only starting from 1st codon position but all the three positions. Thus, addition or deletion of bases taking place during the course of evolution is taken care of. The methodology depicts DNA by condensed matrix counting the rate of presence of adjoining base pairs (Randic, 2000).
Calculation of eigen value and construction of phylogram: Leading eigenvalues were calculated using MATLAB (version 5.0.0.4069) software. Eigenvalues are a special set of scalars associated with a linear system of equations, usually matrix equations that are often regarded as characteristic roots, characteristic values and proper values or latent roots (Mondol et al., 2008). Evaluation of DNA sequences for similarity or dissimilarity is normally aided by the convenience of leading eigenvectors calculated by this method. Diversity between eigenvalues was used to study sequence similarity/dissimilarity keeping in mind the characterization of a sequence by leading eigenvalue (Nandy et al., 2006). Matrices linked to each sequence are estimated and the leading eigen values computed. Variations in leading eigen values concurrent to the string are estimated and the relationships between genes investigated. Distance matrixes of the studied sequences were constructed by summing up the square of the difference of eigen values. Phylograms were built by cluster analysis of the similarity matrix using PHYLIP (Ver 3.65) and drawn with PHYLODRAW (Ver 0.8).
RESULTS AND DISCUSSION
It is seen from the values depicted in Table 1 that the H1N1 viruses, influenza B and C viruses as well as other avian flu viruses are poor GC content genomes with GC values hovering around the 44% mark. This characteristic has been previously reported (Zhou et al., 2005) for H5N1 viruses. Subsequently, their GC3 content is also poor. The GC3 content, which is regarded as an important parameter in studying codon usage variation (Sen et al., 2008), reveals homogeneity in present study. There is very little difference in the values of GC and GC3 content in the individual genes of the genomes (data not shown). Influenza B and C viruses have comparatively poorer GC and GC3 content with respect to H1N1 viruses and avian flu viruses. The Nc values representing the effective number of codons in a gene are quite high. However, the Nc values of the HIN1 CAL/04/2009 strain are much lower compared to other H1N1 strains. Influenza C and B viruses too have lower Nc values compared to other viruses undertaken in this study. The CAI values representing the expression level of the genes are quite high in all the studied genomes with H5N1 goose virus having the highest CAI value of 0.726.
| Table 1: | Mean and standard deviation values of various parameters for the studied viruses |
 | |
Data are expressed as Mean±SD | |
Generally in predicted proteomes the major pI values are classified as belonging to acidic cluster (pI less than 7.4), neutral cluster (pI between 7.4 and 8.1) and basic cluster (pI greater than 8.1) (Nandi et al., 2005). The pI values depicting the isoelectric points of the proteins showed a bi-modal distribution in the studied viral proteomes. All the HIN1 strains as well as Influenza C, H3N2 and H2N2 had a pI in the range greater than 8.1 with H1N1 CAL07/2009 having a pI value of 8.87. On the other hand H5N1, H9N2 and Influenza B had pI values in the neutral cluster. There is good deal of variation in the pI values amongst the proteins in the organisms as exemplified by the standard deviations.
Table 2 shows the correlations of the CAI, GC, GC3, GRAVY, Aromaticity and pI values. The CAI values were correlated with GC and GC3 content (Sen et al., 2008) for the viruses. It was found that CAI showed positive correlations with GC content in all the strains while CAI showed strong positive correlations with GC3 content in some of the strains except H1N1 CAL04/2009, H1N1 CAL05/2009 and H1N1/TEX05/2009.GC3 showed strong correlation with GRAVY representing hydropathicity in case of H1N1 TEX04/2009, H5N1/Goose/Guangdong, H2N2 Korea, H9N2 Hong Kong, Influenza C and H3N2 New York. When GC3 content was correlated with aromaticity values it was found that GC3 showed strong negative correlations with H1N1 CAL04/2009, H1N1 CAL05/2009, H1N1 CAL07/2009 and H1N1 TEX04/2009, respectively. The isoelectric point values were correlated with GC3 and GC content of the viruses. It was noticed that GC3 content had a strong negative correlation with TEX04/2009, H5N1 Goose/Guangdong and positive correlations with H1N1 Puerto Rico, H3N2 New York, Influenza C, H9N2 Hong Kong, H3N2 Korea and H1N1 TEX05/2009. Insignificant correlations were found for the other strains.
Correspondence analysis of codon count (CACC) (Peden, 1999) was computed to infer upon the role of amino-acid compositions in codon usage variations. CACC revealed two major axes of variation. Majority of the genes remained scattered with the exception of Influenza B and C strains were they remain clustered in the centre of the axes. Figure 1a-l show the distribution of the genes along the two major axes of variation. The first major axis of variation was correlated with GC3 content, CAI, Nc, aromaticity and hydropathicity scores.
Figure 2 shows the phylogram constructed for the complete genomes of the studied viruses. The phylogram reveals a rooted tree, which shows two major clades; Clade A and Clade B which contain subclades. Most of the new strains of H1N1 lie together in the same clade with the exception of H1N1 CAL04/2009 that lies in a different clade. The older H1N1 Puerto Rico strain lies in a different clade but in the same major clade with H1N1 CAL04/2009. Among the other viruses Influenza C and Influenza B viruses lie in different clades. H2N2, H5N1, H3N2 and H9N2 lie in same clade. In Clade A it is observed that H1N1 CAL07/2009and H1N1TEX05/2009 co-segregate. Influenza C lie sister to H1N1 strains in Clade A. In Clade B, the Influenza B viruses lie near the H3N2 New York strain. To be specific, these viral strains have more or less similar root distances and remain co-segregated as evident from Fig. 2.
The results obtained for GC3 and GC imply that there is a degree of homogeneity among the genes in the studied viral strains and they are AT rich. Interestingly, earlier reports (Sen et al., 2008) revealed a good deal of heterogeneity in GC rich genomes. Although Nc values varied among the genes in the organisms, high mean Nc values implied low bias. Although, the genomes are rich in AT content yet they are markedly biased. This feature has been previously reported for a Rhizobium phage (Sur et al., 2009). Low bias may be due to the high mutation rates and no contribution from translational selection in influencing codon bias. Comparatively lower Nc values of H1N1 CAL04/2009 and Influenza B and C viruses indicate that they are to some extent different with respect to this feature from other viruses. High CAI values for the studied viruses are quite expected, since all of them are highly pathogenic and need to survive against host defences.
| Table 2: | Correlation results between different indices and principal axis of correspondence analysis on codon count |
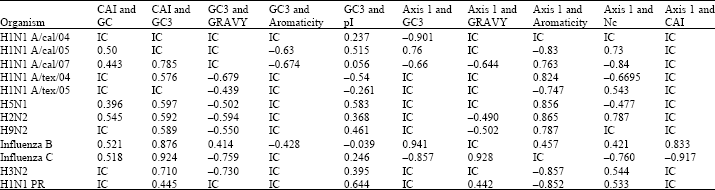 | |
| IC: Inconsequential result | |
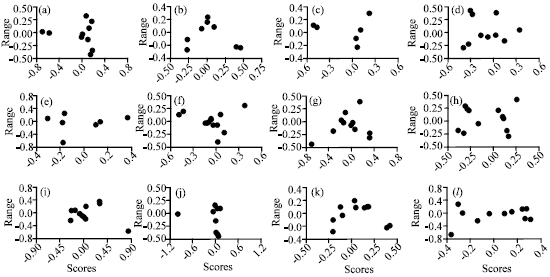 | |
| Fig. 1: | Correspondence analyses of codon count for the studied viral strains. Axis 1 corresponds to X axis and Axis 2 corresponds to Y axis. (a) CAL 04/2009 (b) CAL 05/2009 (c) CAL 07/2009 (d) TEX 04/2009 (e) TEX 05/2009 (f) TEX 05/2009 (g) H5N1/Goose (h) H2N2/Korea (i) H9N1/Honkong (j) Influenza B (k) Influenza C and (l) H1N1/Puertorico |
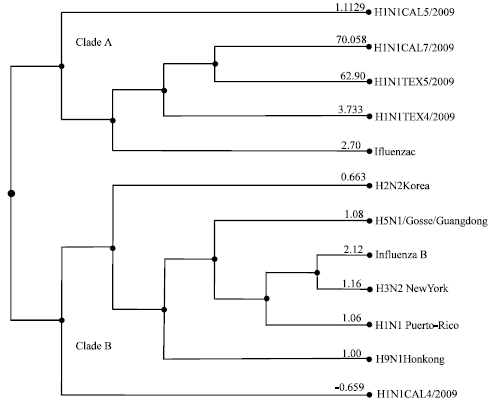 | |
| Fig. 2: | Phylogram of the whole genome segments for the studied viral strains. Numbers depict the root distances |
Most of the genes including hemagglutinin, neuraminidase and structural genes are essential for the survival and manifestation of diseases and the high expression levels of these genes act as a strategy. It is very well known that these viruses have a high rate of mutation in response to drug treatments and the outbreaks that occur after a span of some years (Holmes et al., 2005). The high expressivity of the genes associated with pathogenicity may play a significant role in undergoing reassortment thus giving rise to seasonal outbreaks.
It is shown from Fig. 1 of CACC that there is little difference in variation among the different viruses. However, the clustering of majority of these genes at the centre of the axes for influenza B and C viruses indicate that they may be conserved in nature.
Strong correlations of GC3 and GC content with CAI values point out that expression levels play a significant role in synonymous codon bias in these viruses and GC, GC3 too have an important role to play. Strong correlations of GC3 content with Axis 1 in most of the new H1N1 strains and the two influenza viruses imply that GC compositional content play a significant role in influencing codon bias in these organisms. GC3 content has also been found to show strong negative correlations with grand average hydropathy value (GRAVY) in most of the studied viruses except some of the H1N1 strains. This implies that hydropathy levels increases with the decrease of GC3. Aromaticity levels increase with the decrease in GC3 content as exemplified by strong negative correlations with GC3 in some of the strains. Bimodal distributions of pI values were observed for the viral proteomes. The average pI values for the viruses reveal that the H1N1 strains have more basic proteomes compared to the other viruses. GC3 content shows moderate to low correlations with isoelectric points in the studied viral genomes. Present findings for most of the correlations of GC3 content in the studied viruses strongly support the concept of Kiraga et al. (2007) that low GC rich organisms code for more basic proteomes. However, there have been some exceptions in case of Influenza B, H9N2 and H2N2. Negative correlations of the GC3 content and pI may be attributed to the increase in basic lysine encoded by the AT rich organisms while positive correlations are attributed to the increase in basic arginine. This feature has been previously reported (Kiraga et al., 2007). On the basis of Kiraga et al. (2007) observation, present studied viral proteomes are either basic or neutral, with most of the basic proteomes being influenced by mutational pressure.
Strong correlations of the principal axis of correspondence analysis of Axis 1 with grand average hydropathy value (GRAVY) entails that genes associated with the hydrophilic proteins are favoured by the translationally optimal codons. Correlations of Axis 1 with aromaticity scores point out that aromaticity plays an important role in influencing codon usage patterns in the studied viruses. Similarly gene expression levels strongly influence codon usage bias as exemplified by strong correlations with Axis 1. Negative correlations of the principal axis of variation with Nc values may be attributed to the decrease in codon bias among genes lying left of axis 1, while positive correlations indicate the reverse.
The phylogenetic pattern obtained for the studied viruses using the condensed matrix method point out very clearly that reassortment has a part to play in influencing evolution of these viruses. Clade A and B contained viruses taken on a global basis. Although, most of the H1N1 strains are present together in a single clade two other are placed in a different clade altogether. It is also observed that lineages of one virus are occurring amongst lineages of other viruses. Present results for the whole genome phylogeny reveal that the viral segments being subjected to re-assortments are obtained from various lineages. In this respect our findings support the results of Holmes et al. (2005) that these flu viruses co-circulate, endure and re-assort time-to-time depending upon the environment and susceptibility of the host.
The results obtained from the H1N1 strains commensurate with the aims and objectives of the study. The evolutionary pattern of the new strains has been well explained with the new methodology. The role of mutational pressure as the most important force in guiding the codon usage patterns has been interpreted. Besides, other properties like isoelectric point, aromaticity, hydropathicity, GC content has been shown to influence the lifestyle of the viruses (Tekaia and Yeramian, 2006).
Present results obtained from the condensed matrix based phylogenetic study revealed a slight difference from that obtained by previous studies with respect to the placements of the older H1N1 Puerto-Rico strain and one new strain H1N1 CAL04/2009 that lie in a different clade compared to other new H1N1 strains. Since our methodology focuses on the quantitative as well as qualitative characteristics of the DNA giving equal weightage to all codon positions; the role of mutations in reassortment of these viruses has been interpreted. The cladogram obtained in this study is correct because the result has been further corroborated with the data obtained from isoelectric point with basic proteomes being influenced by mutational pressure.
CONCLUSION
Present findings revealed that synonymous codon usage is less biased in H1N1 virus. Synonymous codon usage study in genes encoded by different influenza A viruses show that they are conserved and mutational bias was the main factor that drives the codon usage variation among these viruses. Low bias may be attributed to the high mutation rates and inability of the contribution of translational selection. High expression of pathogenicity related genes confirm its role as potentially dangerous pathogen. Present studied viral proteomes are either basic or neutral, with most of the H1N1 basic proteomes
influenced by mutational pressure implying the role played by mutations in influencing the nature of the viruses. Genes associated with the hydrophilic proteins are favoured by the translationally optimal codons. Phylogenetic analysis by the condensed matrix method portrays the role played by re-assortments in controlling the evolution of the studied strains. While majority of the new strains lie in the same clade, H1N1 CAL04/2009 lies in the other clade along with H1N1 Puerto Rico. The phylogeny results reaffirm that flu viruses co-circulate and mutate by reassortment depending upon the environment and susceptibility of the host (Holmes et al., 2005).
ACKNOWLEDGMENTS
The authors thank Department of Biotechnology, Government of India for providing financial assistance in setting up Bioinformatics Faculty in the University of North Bengal. A.S. acknowledges the receipt of DBT Overseas Fellowship to University of New Hampshire, USA.
REFERENCES
- Bailly-Bechet, M., M. Vergassola and E. Rocha, 2007. Causes for the intriguing presence of tRNAs in phages. Genome Res., 17: 1486-1495.
PubMedDirect Link - Cox, N.J. and K. Subbarao, 2000. Global epidemiology of influenza: Past and present. Annu. Rev. Med., 51: 407-421.
PubMedDirect Link - Gog, J.R., E.D.S. Afonso, R.M. Dalton, I. Leclercq and L. Tiley et al., 2007. Codon conservation in influenza A virus genome defines RNA packaging signals. Nucl. Acids Res., 35: 1897-1907.
PubMedDirect Link - Knight, C.G., R. Kassen, H. Hebestreit and P.B. Rainey, 2004. Global analysis of predicted proteomes: Functional adaptation of physical properties. Proc. Nat. Acad. Sci. USA., 101: 8390-8395.
PubMed - Lobry, J.R. and C. Gautier, 1994. Hydrophobicity, expressivity and aromaticity are the major trends of amino-acid usage in 999 Escherichia coli chromosome-encoded genes. Nucleic Acids Res., 22: 3174-3180.
PubMed - Markowitz, V.M., N. Ivanova, K. Palaniappan, E. Szeto and F. Korzeniewski et al., 2006. An Experimental metagenome data management and analysis system. Bioinformatics, 22: 359-367.
CrossRef - Mondol, U.K., B. Das, T.C. Ghosh, A. Sen and A.K. Bothra, 2008. Nucleotide triplet based molecular phylogeny of classI and classII aminoacyl t-RNA synthetase in three domain of life process: bacteria, archaea and eukarya. J. Biomol. Struct. Dyn., 26: 321-328.
PubMed - Nandi, S., N. Mehra, A.M. Lynn and A. Bhattacharya, 2005. Comparison of theoretical proteomes: Identification of COGs with conserved and variable pI within the multimodal pI distribution. BMC Genomics, 6: 116-116.
PubMedDirect Link - Nandy, A., M. Harle and S.C. Basak, 2006. Mathematical descriptors of DNA sequences: development and applications ARKIVOC, ix: 211-238.
Direct Link - Randic, M., 2000. Condensed representation of DNA primary sequences. J. Chem. Infrom. Comput. Sci., 40: 50-56.
PubMed - Randic, M., X. Guo and S.C. Basak, 2001. On the characterization of DNA primary sequences by triplet of nucleic acid bases. J. Chem. Inform. Comput. Sci., 41: 619-626.
PubMed - Sen, A., S. Sur, A.K. Bothra, D.R. Benson, P. Normand and L.S. Tisa, 2008. The implication of lifestyle on codon usage patterns and predicted highly expressed genes for three Frankia genomes. Antonie Van Leeuwenhoek, 93: 335-346.
PubMedDirect Link - Sur, S., M. Bhattacharya, A.K. Bothra, L.S. Tisa and A. Sen, 2008. Bioinformatic analysis of codon usage patterns in a free-living diazotroph, Azotobacter vinelandii. Biotechnology, 7: 242-249.
CrossRefDirect Link - Suzuki, Y. and M. Nei, 2002. Origin and Evolution of Influenza virus hemagglutinin genes. Mol. Biol. Evol., 19: 501-509.
PubMedDirect Link - Taubenberger, J.K. and D.M. Morens, 2006. 1918 Influenza: The mother of all pandemics. Emerg. Infect. Dis., 12: 15-22.
CrossRefPubMedDirect Link - Tekaia, F. and E. Yeramian, 2006. Evolution of proteomes: fundamental signatures and global trends in amino acid compositions. BMC Genomics, 7: 307-307.
PubMed - Umashankar, V., V. Arun Kumar and D. Sudarsanam, 2007. ACUA: A software tool for automated codon usage analysis. Bioinformation, 2: 62-63.
PubMed - Webby, R.J. and R.G. Webster, 2003. Are we ready for pandemic influenza?. Science, 302: 1519-1522.
PubMedDirect Link - Wu, G., D.E. Culley and W. Zhang, 2005. Predicted highly expressed genes in the genomes of Streptomyces coelicolor and Streptomyces avermitilis and the implications for their metabolism. Microbiology, 151: 2175-2187.
PubMedDirect Link - Zhou, T., W. Gu, J. Ma, X. Sun and Z. Lu, 2005. Analysis of synonymous codon usage in H5N1 virus and other influenza a viruses. Bio. Syst., 81: 77-86.
PubMedDirect Link
shigha t Reply
Is this H1N1 virus is influensed by anyother viruses
Saubashya Sur
It has been found that HIN1 viral strains are not influenced by any other viruses. We studied the neuraminidase and hemagglitinin genes from H1N1 viruses and found that other viruses do not influence them.