
Satendra Singh
Medicinal Chemistry Division, Dr. B.R. Ambedkar Center for Biomedical Research, University of Delhi, Delhi 110007, India
Pratibha Mehta Luthra
Medicinal Chemistry Division, Dr. B.R. Ambedkar Center for Biomedical Research, University of Delhi, Delhi 110007, India
American Journal of Plant Physiology
Year: 2011 | Volume: 6 | Issue: 4 | Page No.: 228-241







ABSTRACT
In the present study, characterization, isolation and purification of Acid phosphatase from the cotyledons of Psoarlea corylifolia was carried out. The characterization of acid phosphatase includes the optimization of kinetic parameters over the range of substrate concentrations, influence of different divalent metal ions, modulators, various inorganic and organic phosphates on the acid phosphatase mediated hydrolysis of p-Nitrophenyl phosphates. The purification processes included the ammonium sulphate precipitation, DEAE cellulose separation and gel filtration of enzyme through sepharose column. The crude acid phosphatase enzyme showed optimum pH of 5.5 at 37°C in 0.1 M sodium acetate buffer and optimum temperature of 50°C. Km for the substrate p-nitrophenyl Phosphate (p-NPP) was 0.775 mM at optimum pH. Addition of other phosphate substrates such as KH2PO4 and ATP reduced the p-NPP hydrolysis, however, NaH2PO4 and G-6-P did not influence the p-NPP hydrolysis. CuSO4, Sodium flouride (NaF) and Sodium molybdate (NaMoO4) strongly inhibited the acid phosphatase activity. Partially purified acid phosphatase protein (226.08 fold) obtained by ammonium sulphate precipitation and gel filtration exhibited very high specific activity (264211.157 μmole min-1 mg-1). Acid phosphatase showed spots at ~28 kDa and ~30 kDa in two dimensional gel electrophoresis followed by western blot analysis.
PDF Abstract XML References Citation
Received: March 03, 2011;
Accepted: July 02, 2011;
Published: August 01, 2011
How to cite this article
Satendra Singh and Pratibha Mehta Luthra, 2011. Identification, Characterization and Partial Purification of Acid Phosphatase from Cotyledons of Psoralea corylifolia L. American Journal of Plant Physiology, 6: 228-241.
DOI: 10.3923/ajpp.2011.228.241
URL: https://scialert.net/abstract/?doi=ajpp.2011.228.241
DOI: 10.3923/ajpp.2011.228.241
URL: https://scialert.net/abstract/?doi=ajpp.2011.228.241
INTRODUCTION
Psoralea corylifolia L. (Bhavanchi, Fabaceae), an annual herb distributed widely in tropical and sub-tropical regions, exhibits properties such as antitumor, antibacterial, antifungal and antioxidative activities and is used in curing stomach ache, anthelmintic, diuretic and diaphoretic in febrile conditions (Baskaran and Jayabalan, 2007; Chanda et al., 2011). The active principle of the plant consist commercially valuable psoralen and isopsoralen. Psoralen is used for the photochemotherapy of vitiligo and skin diseases such as psoriasis, mycosis fungoides and eczema due to photosensitizing, photobiological and phototherapeutic properties (Frank et al., 1998). Wild population of Psoralea has declined due to poor rate of seed germination (Baskaran and Jayabalan, 2008, 2009), a process that requires acid phosphatase enzyme to drive metabolic processes in the cell. Acid phosphatase catalyzes the hydrolysis of tLuthrahe terminal phosphate of phosphomonoesters to release inorganic phosphate and free energy required for seed germination (Guo and Pesacreta, 1997). The hydrolysis of phosphomonoesters by phosphatases in biological systems is an important process. This process is linked to energy metabolism, metabolic regulation and a wide variety of cellular signal transduction pathways. The role of acid phosphatase in phosphorus metabolism has been extensively studied in prokaryotic and eukaryotic systems (Al-Omair, 2010). Plants use the enzyme to scavenge phosphate from organic sources under phosphate-limited conditions (Lefebvre et al., 1990), pathogen and salt stress (Parida and Das, 2004; Schweighofer et al., 2004) and development (Luan, 1998). Plant acid phosphatase contributes to the mobilization of phosphate from macromolecular organic phosphates during seed germination and seedling growth (Duff et al., 1994). The functioning of different metabolic processes and growth of seedlings during germination are essentially dependent on the acid phosphatase catalyzed solubilization and mobilization of organic phosphates in soil (Bishnoi et al., 1993; Kawarasaki et al., 1996).
In the present investigation, we have characterized the properties of acid phosphatase enzyme from the crude extract of cotyledons of medicinally valuable Psoralea corylifolia for the first time as a step towards understanding its properties. The optimum concentration of substrate p-NPP, pH, temperature and influence divalent metal ions, modulators and some of the phosphate substrate were determined. Further, the enzyme was partially purified and was analyzed by two dimensional gel electrophoresis followed by western blot.
MATERIALS AND METHODS
Materials: Seeds of Psoralea were obtained from local market of Delhi, India. p-NPP (p-Nitrophenyl phosphate), p-NP (p-Nitrophenol), NaF, NaMoO4, ATP, G-6-P, KH2PO4, NaH2PO4, BSA (bovine serum albumin), Coomassie brilliant blue G-250 and Phenylmethylsulphonyl flouride (PMSF) were purchased from Sigma Chemical Company (St. Louis, MO, USA). The HPLC solvents and chemicals were purchased from E-Merck.
Germination of Psoralea seeds: Seeds of Psoralea were surface sterilized with 2% NaClO solution for 10 min, washed with sterile water and germinated in autoclaved vermiculite in dark at 30°C incubator in July 2009. After 48 h of germination, plants were harvested, uprooted and thoroughly rinsed with distilled water. Cotyledons and embryonic axis were carefully detached, rinsed with distilled water, dried with filter paper, weighed and stored at -80°C.
Extraction of acid phosphatase: Cotyledons and embryonic axis (1 g) were ground separately in a mortar (1 g 9 mL-1 buffer) with an extraction mixture consisting of cold 0.1 M K2HPO4/KH2PO4 (K-Pi) buffer (pH 5.5), 2% nonyl phenyl ethylene glycol (NP- 40), 1 μg mL-1 lysozyme 1 μg mL-1 each of protease inhibitor cocktail (pepstatin, leupeptin and apoprotinin) along with 10 mM Phenyl Methyl Sulphonyl Fluoride (PMSF) to give separate homogenous mixture of cotyledons and embryonic axis (Luthra and Singh, 2010). Each homogenate was centrifuged at 17000 rpm at 4°C for 20 min, producing a supernatant designed as crude extract.
Assay
Colorimetry assay for acid phosphatase: Activity of acid phosphatase (crude extract) was assayed at 37°C. The assay started after adding 15 μL of total homogenate to reaction mixture (985 μL) containing 2 mM p-NPP in 0.1 M sodium acetate buffer (pH 5.5). After 60 min the reaction was stopped by adding 100 μL of 2 N NaOH; the absorbance of the solution was measured at 405 nm using a Multimode microplate reader (Biotek Synergy, USA) (Senna et al., 2006).
| Table 1: | Epsilon values of p-NP at different pH |
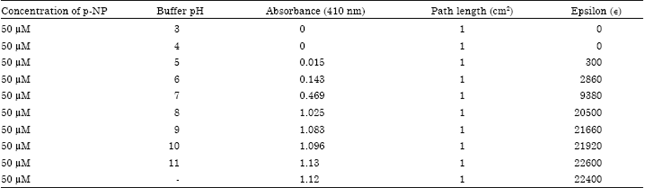 | |
| - Reach Buffer stopped by 2N NaoH | |
The results were expressed in μ mole min-1 mg-1, where μ mole represents the one International Unit of enzyme (One unit is defined as the amount of the enzyme that catalyzes the conversion of 1 μ mole of substrate per minute). Zero time blank containing all reagents, except plant extract, was always included in the assay. The linearity was evaluated by determining standard working solution containing various concentration of p-NP. The activity of acid phosphatase was measured in μ mole min-1 mg-1 protein versus p-NPP hydrolysis. Absorbance and concentration were subjected to least square linear regression and correlation co-efficient. Molar extinction coefficient of p-NP was measured at different pH to correlate the data obtained by colorimetric assay (Table 1).
HPLC: Samples of acid phosphatase catalyzed reaction were also analyzed by HPLC (model series 200 pump; Perkin Elmer) using C18 column (ODS, 250x4.8 mm, 5 μm; Hypersil). The mobile phase consisted of sodium acetate buffer (0.1 M, pH 5.5; at a flow rate of 1 mL min-1). The sample (20 μL) was detected using UV/vis detector model 200 (Perkin Elmer) at λ max 320 nm. The retention time (Rt) of p-NPP and p-NP were at 2.3 min and 18.0 min, respectively.
Kinetic characterization
Influence of pH: Total homogenate (15 μL) from cotyledons was used to carry the pH optimum study for the enzyme. Various buffers were prepared at different pH such as 0.1 M sodium citrate (pH 3, 4), 0.1 M sodium acetate (pH 5.0, 5.5, 6.0), 0.1 M Tris (pH 7.0) and 0.1 M glycine (pH 8.0). p-NPP (2 mM) was used as a substrate for measuring the pH optimum at 37°C.
Effect of temperature: The optimum temperature for maximum acid phosphatase activity was determined by measuring the enzyme activity at various temperatures (10, 20, 30, 40, 50, 60, 70 and 80°C). The enzyme reaction buffer containing 2 mM p-NPP was incubated for 5 min at each temperature. The supernatant from cotyledons was then added to measure the enzyme activity.
Influence of various metal ions and modulators: The influence of metal ions (MnO2, NiCl2, FeCl2, 7 H2O, CoCl2 6H2O, ZnSO4, MgSO4 7H2O, CuSO4 5H2O) and modulators (NaMoO4 and NaF) on catalytic activity of acid phosphatase was assessed by adding different concentrations (0-2000 μM and 0-16 mM, respectively) of metal/modulator to the enzymatic reaction. The mixture was incubated at 37°C for 60 min. The results were expressed as relative activity in respect to the control reaction without metal/modulator added.
Influence of inorganic and organic phosphates on p-NPP hydrolysis: Various inorganic (NaH2PO4 and KH2PO4) and organic (ATP and G-6-P) phosphates (0-16 mM) were used along with 2 mM p-NPP to assess their influence on p-NPP hydrolysis. The reduction in p-NPP hydrolysis was measured in the presence of inorganic/organic phosphates and compared to control containing 2 mM of p-NPP only.
Isolation and purification of acid phosphatase: All procedures were carried out at 4°C. Enzyme activity was determined using the ELISA plate reader. K-Pi buffer (section 2.3) was used throughout the purification process.
Ammonium sulphate precipitation: The crude extract from cotyledons was brought to 30% saturation with ammonium sulphate in an ice bath under slow stirring for 1 h and centrifuged at 5000 rpm for 15 min. The treatment was repeated to give a final saturation of 90%. The saturated (90%) extract was centrifuged and the supernatant was discarded. The 20 fold active pellet was re-suspended in the extraction buffer and retained for further purification.
Chromatographic purification: The re-suspended pellet in extraction buffer (2 mL) was loaded on a sepharose 6 B column (2.5x10 cm) equilibrated with same buffer. The enzyme was eluted at a flow rate of 40 mL h-1. Fractions of 2 mL were collected and the fractions with acid phosphatase activity (colorimetric assay) were retained for further analysis.
Two dimensional gel electrophoresis: Active fractions obtained after partial purification of the enzyme (226.08 fold) were treated with ice-cold acetone (1:5 v/v) at -20°C overnight, centrifuged at 4°C, 5000 rpm for 15 min to precipitate the proteins. K-Pi buffer (0.1 M, pH 5.5) with protease inhibitor cocktail (1 μg mL-1) was used to dissolve the proteins and dialyzed overnight against water. The dialyzed protein sample was centrifuged at 15000 rpm for 10 min. The precipitated proteins were washed with ice-cold acetone containing 0.07% 2-mercaptoethanol to remove pigments and lipids until the supernatant was colorless. The pellet was vacuum-dried, re-suspended in re-solubilization buffer, followed by sonication (10 secx3 cyclex50% power) and centrifugation (15000 rpm for 20 min).
The pellet obtained from centrifugation was dissolved in re-hydration buffer (Dong et al., 2009) and IEF (Protein IEF Cell BIO-RAD) was performed as previously reported by Xu et al. (2008) on a 11 cm immobilized pH (3-10) strip. Focused proteins and pre-stained broad range protein markers (New England Lab) were resolved by SDS-PAGE using 12% (w/v) of acrylamide. Gel was stained with colloidal coomassie blue. Western blot analysis of partially purified protein were carried out with potato polyclonal antibody as described previously (Abdelmeguid et al., 2008).
Protein quantification: Total protein concentration was determined by the 2-D quanta kit (GE Amersham Biosciences).
Statistical analysis: The Mean±S.E.M. was expressed for values obtained from a minimum of three experiments. Data were analyzed by one-way ANOVA; post method student’s t test (Graph pad Prism Software). Differences with a p-value less than 0.05 were considered significant.
RESULTS
Acid phosphatase activity in cotyledons and embryonic axis of Psoralea: Enzyme activity was evaluated using p-NPP as synthetic substrate in cotyledons and embryonic axis of Psoralea seedlings (Fig. 1). Variation in enzyme activity was observed in cotyledons and embryonic axis of Psoralea. Crude extract from cotyledons showed 2.5 fold higher acid phosphatase activity (2412±35.86 μ mole min-1 mg-1) as compared with activity observed in the crude extract from embryonic axis (1028±40.71 μ mole min-1 mg-1 Fig. 1).
HPLC quantification of acid phosphatase catalyzed p-Nitrophenol synthesis: HPLC quantification of acid phosphatase induced (catalyzed) hydrolysis of p-NPP (2 mM; Rt 2.3 min) showed that the formation of 880 μM p-NP (Rt 18.0 min) after 60 min (Fig. 2a) However, conversion of p-NPP (2 mM) to p-NP was very low (138 μM) after 10 min of incubation (Fig. 2b).
Measurement of Km and Vmax for acid phosphatase: The rate of hydrolysis of the substrate p-NPP (0.015-8 mM) in the presence of Psoralea acid phosphatase showed that optimum concentration of p-NPP was 8 mM for acid phosphatase catalyzed hydrolysis in to p-NP (Fig. 3a); thereafter stability in hydrolysis was observed. The apparent Km value for p-NPP was 0.775 mM and the maximum reaction rate (Vmax) was 3.48 x 103 μ mole min-1 mg-1 protein at pH 5.5 (Fig. 3b).
Influence of pH: The effect of pH (Fig. 4a) was determined with crude extract using p-NPP as a substrate, the enzyme showed significant acid phosphatase activity in the range of pH 4.0-5.5, with maximal activity at pH 5.5. Lower than 4.0 pH and upper than 5.5 pH value strongly inhibited the activity of the enzyme.
To evaluate whether lower (3.0) and higher (8.0) pH range inhibited or inactivated the enzyme; the enzyme was incubated with p-NPP at these pH for 1 h. The reaction mixture with either lower or higher pH range was separately brought to optimal pH. p-NPP hydrolysis was not observed at optimal pH after 1 h incubation of the reaction mixture. Addition of fresh crude extract to the above reaction mixture showed normal enzyme activity.
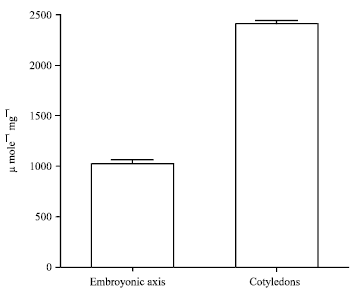 | |
| Fig. 1: | Acid phosphatase activity of germinating seedlings of Psoralea corylifolia a. Seedlings after 48 h of germination in dark showing cotyledons and embryonic axis. Specific activity of acid phosphatase in cotyledons and embryonic axis. The results given are Mean±S.E.M; n = 3 |
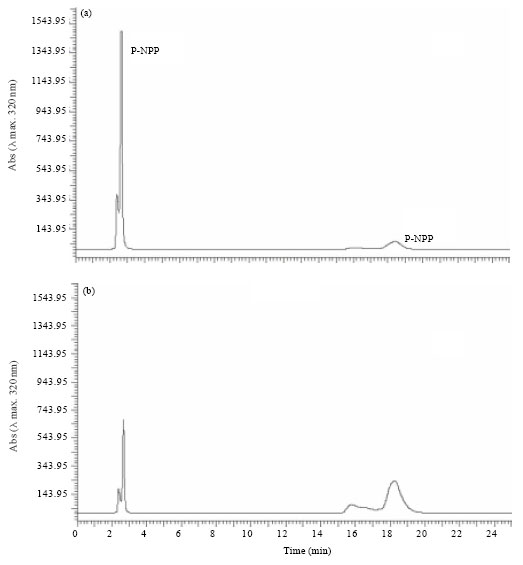 | |
| Fig. 2(a-b): | HPLC chromatograms of acid phosphatase catalyzed dephosphorylation of p-NPP (flow rate 1 mL min-1, λmax 320 nm). (a) Reaction mixture and enzyme extract with 2 mM of p-NPP after 10 min of incubation (p-NPP, Rt 2.3 min; p-NP, Rt 18.0 min) and (b) after 60 min of incubation |
These results indicated that high or low pH of medium inactivated the acid phosphatase enzyme from Psoralea.
In the time course study, the maximum de-phosphorylation of p-NPP (2613±8.8 μ mole min-1 mg-1) was observed after 90 min of incubation and remained constant in extended time intervals (Fig. 4b).
Effect of temperature on the enzyme activity: The optimum temperature for maximum acid phosphatase activity was determined by measuring the enzyme activity at various temperatures from 0 to 80°C. The optimum temperature for de-phosphorylation of p-NPP was found to be 50°C with catalytic activity of 8297±4655 μ mole min-1 mg-1. Acid phosphatase was quite stable at higher temperature range of 70 to 80°C with significant catalytic activity of 5790±48.60 and 4852±37.27 μ mole min-1 mg-1, respectively (Fig. 5).
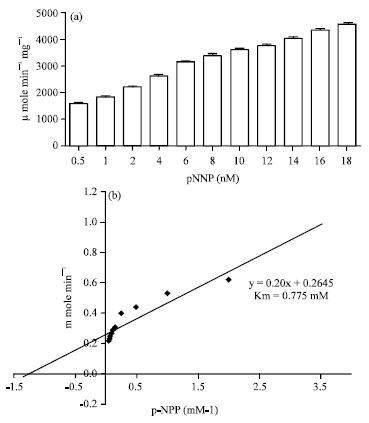 | |
| Fig. 3(a-b): | Kinetic constant of acid phosphatase with p-NPP from cotyledons of Psoralea. (a) Gradual increase in activity of acid phosphatase was observed over a range of substrate concentrations. (b) Line-weaver Burk plot was used to calculate the kinetic constants (Km of 0.775 mM and a Vmax of 3.48x103 μ mole min-1 mg-1). The results given are Mean±SE (n = 3) |
Influence of metal ions on acid phosphatase activity: The effect of metal ions (Mg++, Co++, Zn++, Ni++, Mn++, Fe++ and Cu++) as possible activators or inhibitors of acid phosphatase showed that Mg++ was observed least (2175.34±5.6 μ mole min-1 nkat mg-1) and Cu++ most (452.6±2.6 μ mole min-1 nkat mg-1) toxic metal (at 2000 μM) as compared to control (2419.61±11.73 μ mole min-1 nkat mg-1 Table 2). The maximum inhibition in enzyme activity was observed at the concentration of 2000 μM of metal in the order of Cu++>Zn++>Ni++>Fe++>Co++>Mn++> Mg++.
Influence of modulators: Influence of two important modulators NaF and NaMoO4 on acid phosphatase was studied. NaF influenced the activity of acid phosphatase in dose dependent manner. Gradual reduction in p-NPP hydrolysis was observed with increasing concentrations of NaF and at lower concentration of NaF, no significance inhibition was observed. Lower concentration of NaMoO4 (0.03 mM) strongly reduced the activity of acid phosphatase from cotyledons of Psoralea. The results demonstrate that NaMoO4 is a strong inhibitor of acid phosphatase extracted from cotyledons of Psoralea (Fig. 6).
Substrate specificity of Psoralea acid phosphatase: The substrate specificity of acid phosphatase was tested with various potential competitive substrates with 2mM p-NPP at a concentration of 0.03-16 mM by measuring the reduction in p-NPP hydrolysis. A gradual reduction in p-NPP hydrolysis was observed with the increasing concentration of inorganic or organic phosphates (Table 3).
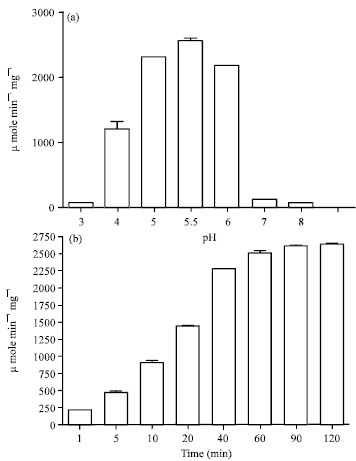 | |
| Fig. 4(a-b): | Effect of pH and time course study of Psoralea acid phosphatase of (a).To ascertain the pH optima of the crude enzyme, p-NPP hydrolysis activity was measured at different pH varying from 3-8 in different buffers (0.1 M) H2PO4/K2HPO4 (K-Pi), sodium acetate, glycine, Tris-HCl and sodium citrate at 37°C under standard assay condition (b). To assess the time course study, after a specific time interval, the reaction was stopped with 100 μL of 2 N NaOH; total p-NP formed was read at 405 nm. The results given are Mean±S.E.M (n = 3) |
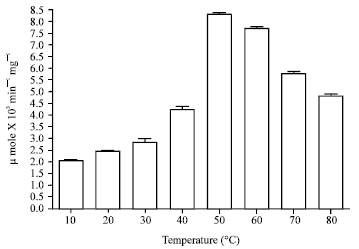 | |
| Fig. 5: | Thermal stability of acid phosphatase from crude extract of cotyledons of Psoralea corylifolia. The crude extracts from cotyledons were incubated at 10-80°C for 60 min. The residual enzyme activity was analyzed by colorimetric assay. The results given are Mean±S.E.M (n = 3) |
| Table 2: | Effect of divalent metal ions on in vitro acid phosphatase activity |
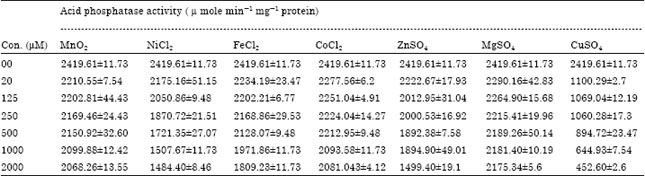 | |
| aEach value represents the average and standard deviation (student’s t test) | |
| Table 3: | Effect of competitive substrates on p-NPP hydrolysis |
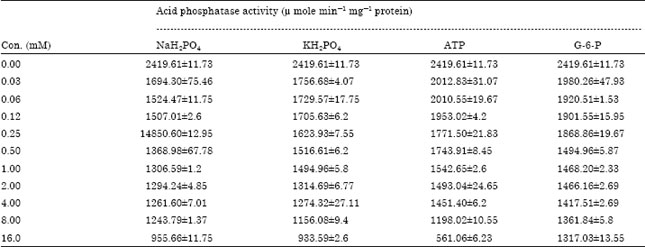 | |
| aConditions are described in materials and methods. Each value represents the average and standard deviation (student’s t test) | |
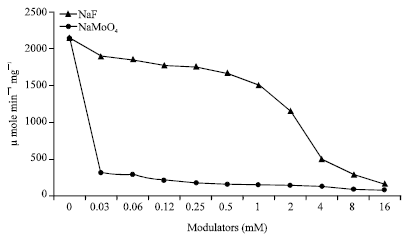 | |
| Fig. 6: | Effect of modulators on acid phosphatase activity from crude extract of cotyledons of Psoralea. The results given are Mean±SEM (n = 3) |
The results established that acid phosphatase in Psoralea cotyledons was non-specific.
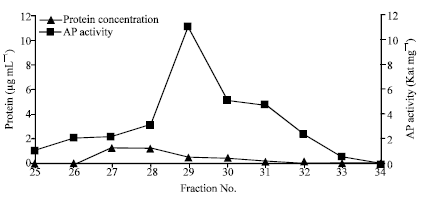 | |
| Fig. 7: | Purification profile of Psoralea cotyledon acid phosphatase activities and protein concentration through Sepharose 6B column chromatography |
| Table 4: | Summary of purification procedure of acid phosphatase from cotyledons (3 g) of Psoralea |
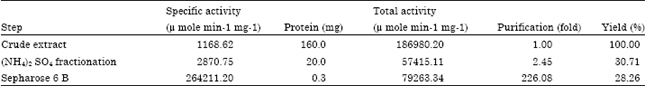 | |
Isolation and purification of acid phosphatase: The cotyledons were used for the purification of enzyme because cotyledons showed higher activity of acid phosphatase as compared to embryonic axis. Ammonium sulphate precipitation and chromatographic purification of crude extract from cotyledons of Psoralea gave partially purified (226.08 fold) enzyme as a single peak with a yield of 28.26% (Fig. 7). The specific activity of the partially purified acid phosphatase was 264211.157 μmole min-1 mg-1 of protein. The results of the purification are summarized in Table 4.
Two Dimensional gel electrophoresis and Western-blot analysis of partially purified acid phosphatase: The iso-electric focusing and SDS-PAGE analysis of the single peak obtained from gel filtration showed partially purified acid phosphatase protein (Fig. 8a). The focused proteins on Western blot analysis displayed five major protein spots with a molecular mass of ~28 and ~30 kDa, respectively (Fig. 8b).
DISCUSSION
The acid phosphatase maintains an adequate phosphate level for seed germination and enzyme activity in the cells which is directly related to the phosphate content (Prazeres et al., 2004). We observed very high acid phosphatase activity in the cotyledons of germinating seedlings of Psoralea as compared to embryonic axis. Increased acid phosphatase activity could be related to either de novo protein synthesis or to the activation of a pre-existing protein (Garcia et al., 2004).
The acid phosphate activity has been reported in several plant species and large variation in activity was found in different plant parts (Panara et al., 1990; Waymack and Van Etten, 1991; Duff et al., 1991; LeBansky et al., 1991; Ullah and Gibson, 1988; Penheiter et al., 1997). Psoralea cotyledons showed relatively higher Km (0.775 mM) at pH 5.5 from crude extract. The plants (Glycine max, Phaseolus vulgaris, Triticum vulgare and Zea maize) generally with high rate of germination possessed low Km higher Km for the acid phosphatase for could be responsible for poor rate of seed germination in Psoralea.
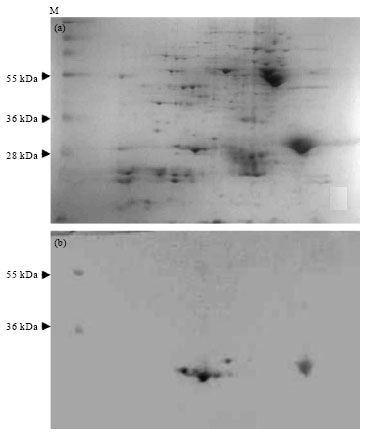 | |
| Fig. 8(a-b): | 2-D gel electrophoresis and western blot of partially purified acid phosphatase from cotyledons of Psoralea. (a) Coomassie blue-stained protein, (200 μg) was loaded on a 11 cm (pH 3-10) immobiline dry strip for IEF followed by SDS-PAGE (12 %). Pre-stained broad range protein markers were used for SDS-PAGE. (b) Immunoblot of total protein from cotyledons probed with purified polyclonal potato acid phosphatase antibody reacted with five spots of ~28 kDa and 30 kDa protein bands of acid phosphatase using potato acid phosphatase polyclonal IgG antibody |
In general, the optimum pH value for acid phosphatase has been found to be in pH range 3-6 (Juma and Tabatabai, 1988), the optimum pH for acid phosphate extracted from cotyledons of Psoralea was 5.5.
The enzyme displayed significant activity in narrow pH range (4.0-5.5) i.e., below pH 4.0 and above pH 5.5, the activity diminished strongly and 80% reduction in p-NPP hydrolysis was observed at pH 6.0. Acid phosphatase from Psoralea was quite stable to heat demonstrating significant activity at 80°C, however, possessed maximum activity at 50°C. Similar result of acid phosphatase activity was observed by Matinizadeh et al. (2008). Heat stable acid phosphatase has been reported in several plants such as bean (Garcia et al., 2004), banana (Zhou et al., 2003), tomato and lupin roots (Li and Tadano, 1996).
Enzyme reactions are inhibited by metals which may form complex with the substrate, or combine with the protein-active groups of the enzymes, or react with the enzyme-substrate complex (Hinajosa et al., 2004). Acid phosphatase activity of wheat roots was inhibited in the presence of Cu++, Fe+++, Zn++ and Co++, respectively (Hasegawa et al., 1976). The metals Cu++, Fe+++ and Zn++, inhibited the acid phosphatase activity of White Lupine seedlings (Newmark and Wenger, 1960) and tobacco leaves (Roberts, 1956). Present results showed significant inhibition of the acid phosphatase activity from Psoralea cotyledons in the presence of divalent metal cations in the order of Cu++>Zn++>Ni ++>Fe++>Co++>Mn++>Mg++. NaF and NaMoO4 are strong inhibitors of the acid phosphatase (Senna et al., 2006; Smith and Walker, 1996; Van Etten et al., 1974), similar results were found in Psoralea acid phosphatase. Although, NaMoO4 inhibited the enzyme at all concentrations but NaF did not influence the enzyme activity at lower concentrations (Fig. 6). Acid phosphatase from Psoralea has been found to be non-specific. KH2PO4 and ATP reduced the hydrolysis of p-NPP to 60% at maximum concentration of 16 mM, however, NaH2PO4 and G-6-P did not affect the hydrolysis of synthetic substrate p-NPP.
Low acid phosphatase activity has been reported in several plants such as 418-nkat mg-1 (25.07 μmole min-1 mg-1) protein from Barley coleoptiles (Pasqualini et al., 1997), 1500 nkat mg-1 (89.98 μmole min-1 mg-1) from apple leaves (Zhang and McManus, 2000) however, acid phosphatase purified from roots of white clover (Cirkovic et al., 2002) showed reasonably good activity (9000 nkat mg-1 protein or 539.89 μmole min-1 mg-1). Crude extract from cotyledons of Psoralea possessed low enzyme activity (2412±35.86 μmole min-1 mg-1) however, high specific activity (264211.157 μmole min-1 mg-1 protein) was found in partially purified protein. The isoforms of acid phosphatase in root nodule of P. vulgaris (Garcia et al., 2004) and pollen extract of Artemisia vulgaris (Cirkovic et al., 2002) have been reported previously. In western blot analysis of partially purified acid phosphatase from cotyledons of Psoralea, the spots with a molecular mass of ~28 kDa and 30 kDa could be isoforms of the same enzyme (Fig. 8b).
CONCLUSION
In conclusion, the acid phosphatase from crude extract of cotyledons of Psoralea has been found to possess relatively low affinity for the substrate which may be responsible for poor rate of seed germination. The enzyme activity was strongly inhibited by divalent metal Cu++ and NaMoO4 at low concentrations. Further, characterization of the isoforms from the purified enzyme will provide a scaffold leading to define its role during seed development.
ACKNOWLEDGMENTS
Satendra Singh is thankful to University Grant Commission for financial support, New Delhi, India.
REFERENCES
- Abdelmeguid, N.E., M.H. Mostafa, A.M. Abdel-Moneim, A.F. Badawi and N.S. Abou Zeinab, 2008. Tamoxifen and melatonin differentially influence apoptosis of normal mammary gland cells: Ultrastructural evidence and p53 expression. Int. J. Cancer Res., 4: 81-91.
CrossRefDirect Link - Al-Omair, M.A., 2010. Purification and biochemical characterization of acid phosphatase from Vigna aconitifolia. Am. J. Plant Physiol., 5: 361-370.
CrossRef - Baskaran, P. and N. Jayabalan, 2007. Rapid micropropagation of Psoralea corylifolia L. using nodal explants cultured in organic additive-supplemented medium. J. Hort. Sci. Biotechnol., 82: 908-913.
Direct Link - Baskaran, P. and N. Jayabalan, 2009. In vitro propagation of Psoralea corylifolia L. by somatic embryogenesis in cell suspension culture. Acta Physiol. Plant., 31: 1119-1127.
CrossRef - Bishnoi, N.R., I.S. Sheoran and R. Singh, 1993. Effect of cadmium and nickel on mobilisation of food reserves and activities of hydrolytic enzymes in germinating pigeon pea seeds. Biol. Planta, 35: 583-589.
CrossRefDirect Link - Chanda, S., M. Kaneria and R. Nair, 2011. Antibacterial activity of Psoralea corylifolia L. seed and aerial parts with various extraction methods. Res. J. Microbiol., 60: 124-131.
CrossRefDirect Link - Xu, C., X. Yan and B. Huang, 2008. Protein extraction for two-dimensional gel electrophoresis of proteomic profiling in turfgrass. Crop Sci., 48: 1608-1614.
Direct Link - Cirkovic, T.D., D.J. Marija, M. Gavrovic-Jankulovic, J.L. Bukilica Mandic, S.Z. Petrovic and R.M. Jankov, 2002. Isolation and partial characterization of an acid phosphatase from Artemisa vulgaris pollen extract. J. Serb. Chem. Soc., 67: 567-572.
Direct Link - Dong, H.X., H.X. Li, G.S. Xie and H.L. Zeng, 2009. Identification of differentially expressed proteins associated with chlorophyll-deficient mutant rice. Asian J. Plant Sci., 8: 344-352.
CrossRefDirect Link - Duff, S.M.G., D.D. Lefebvre and W.C. Plaxton, 1991. Purification, characterization, and subcellular localization of an acid phosphatase from black mustard cell-suspension cultures: Comparison with phosphoenolpyruvate phosphatase. Arch. Biochem. Biophys., 286: 226-232.
CrossRef - Duff, S.M.G., G. Sarath and W.C. Plaxton, 1994. The role of acid phosphatases in plant phosphorus metabolism. Physiol. Plant., 90: 791-800.
CrossRefDirect Link - Frank, S., S. Caffieri, A. Raffaelli, D. Vedaldi and F. Dall'Acqua, 1998. Characterization of psoralen-oleic acid cycloadducts and their possible involvement in membrane photodamage. J. Photochem. Photobiol. B., 44: 39-44.
CrossRef - Garcia, N.A.T., M. Olivera, C. Iribarne and C. Lluch, 2004. Partial purification and characterization of a non-specific acid phosphatase in leaves and root nodules of Phaseolus vulgaris. Plant Physiol. Biochem., 42: 585-591.
CrossRef - Guo, J. and T.C. Pesacreta, 1997. Purification and characterization of an acid phosphatase from the bulb of Allium cepa L. var. sweet Spanish. J. Plant Physiol., 151: 520-527.
Direct Link - Hasegawa, Y., K.R. Lynn and W.J. Brockbank, 1976. Isolation and partial characterization of cytoplasmic and wall-bound acid phosphatase from wheat roots. Can. J. Bot., 54: 1163-1169.
Direct Link - Hinajosa, B.M., J.A. Carreira, R. Garcia-Ruiz and P.R. Dick, 2004. Soil moisture pre-treatment effects on enzyme activities as indicators of heavy metal-contaminated and reclaimed soils. Soil Biol. Biochem., 36: 1559-1568.
CrossRefDirect Link - Juma, N.G. and M.A. Tabatabai, 1988. Phosphatase activity in corn and soybean roots: Conditions for assay and effects of metals. Plant Soil, 107: 39-47.
CrossRef - Kawarasaki, Y., H. Nakano and T. Yamane, 1996. Purification and some properties of wheat germ acid phosphatases. Plant Sci., 119: 67-77.
CrossRefDirect Link - LeBansky, B.R., T.D. McKnight and L.R. Griffing, 1991. Purification and characterization of a secreted purple phosphatase from soybean suspension cultures. Plant Physiol., 99: 391-395.
PubMedDirect Link - Lefebvre, D.D., S.M. Duff, C.A. Fife, C. Julien-Inalsingh and W.C. Plaxton, 1990. Response to phosphate deprivation in Brassica nigra suspension cell, enhancement of intracellular cell surface and secreted in Pi-absorption rate. Plant Physiol., 93: 504-511.
PubMed - Li, M. and T. Tadano, 1996. Comparison of characteristics of acid phosphatases secreted from roots of lupin and tomato. Soil Sci. Plant Nutr., 42: 753-763.
Direct Link - Luan, S., 1998. Protein phosphatases and signaling cascades in higher plants. Trends Plant Sci., 3: 271-275.
CrossRefDirect Link - Matinizadeh, M., S.A.A. Korori, M. Teimouri and W. Praznik, 2008. Enzyme activities in undisturbed and disturbed forest soils under oak (Quercus brantii var. persica) as affected by soil depth and seasonal variation. Asian J. Plant Sci., 7: 368-374.
CrossRefDirect Link - Panara, F., S. Pasqualini and M. Antonielli, 1990. Multiple forms of barley root acid phosphatase: purification and some characteristics of the major cytoplasmic isoenzyme. Biochim. Biophys. Acta, 1037: 73-80.
PubMedDirect Link - Pasqualini, S., F. Panara, L. Ederli, P. Batini and M. Antonielli, 1997. Multiple acid phosphatase in barley coleoptiles. Isolation and partial characterization of the 63 kDa soluble enzyme form. Plant Physiol. Biochem., 35: 95-101.
Direct Link - Penheiter, A.R., S.M.G. Duff and G. Sarath, 1997. Soybean root nodule acid phosphatase. Plant Physiol., 114: 597-604.
Direct Link - Prazeres, J.N., C.V. Ferreira and H. Aoyama, 2004. Acid phosphatase activities during the germination of Glycine max seeds. Plant Physiol. Biochem., 42: 15-20.
Direct Link - Roberts, D.W., 1956. The wheat leaf phosphatases. I. A survey of the inhibitors at pH 5.7. J. Biol. Chem., 219: 711-718.
PubMed - Smith, R.D. and J.C. Walker, 1996. Plant protein phosphatases. Annu. Rev. Plant Physiol. Plant Mol. Biol., 47: 101-125.
Direct Link - Zhang, C. and M.T. McManus, 2000. Identification and characterisation of two distinct acid phosphatases in cell walls of roots of white clover. Plant Physiol. Biochem., 38: 259-270.
CrossRef - Zhou, R.L., L. Cheng and R.O. Wayne, 2003. Purification and characterization of sorbitol-6-phosphate phosphatase from apple leaves. Plant Sci., 165: 227-232.
CrossRefDirect Link - Baskaran, P. and N. Jayabalan, 2008. Effect of growth regulators on rapid micropropagation and psoralen production in Psoralea corylifolia L. Acta Physiol. Plant, 30: 345-351.
CrossRef - Luthra, P.M. and S. Singh, 2010. Identification and optimization of tyrosine hydroxylase activity in Mucuna pruriens DC. var. utilis. Planta, 1: 1361-1369.
CrossRefPubMedDirect Link