
Ravindra B. Malabadi
Division of Plant Biotechnology, Department of Botany, Karnatak University, Pavate Nagar, Dharwad-580003, Karnataka State, India
K. Nataraja
Division of Plant Biotechnology, Department of Botany, Karnatak University, Pavate Nagar, Dharwad-580003, Karnataka State, India
American Journal of Plant Physiology
Year: 2007 | Volume: 2 | Issue: 6 | Page No.: 333-343







ABSTRACT
Present research highlights for the first time the expression of cDNA clones of genes involved in programming the apical meristem cells towards somatic embryogenic pathway influenced by external environmental stimulus like cold-pretreatment. Differential display was used to isolate the genes which are expressed specifically in embryogenic tissue induced by cold-pretreatment of thin sections of vegetative shoot apices of mature trees of Pinus roxburghii. We have developed a rapid method which employs magnetic beads to capture mRNA from cell lysates. Of the 56 cold-enhanced embryogenic-associated cDNAs identified, 20 were cloned. Nine of the 20 fragments which generated single bands on re-amplification were selected for cloning and further analysis. During reverse northern hybridization, all the 20 clones selected generated a positive signal when probed with labeled cDNA from cold-enhanced embryogenic tissue, but no signal when probed with cDNA from the non-embryogenic tissue (control treatment). All the 20 clones thus contained inserts that were specific to cold-enhanced somatic embryogenesis. This approach allows us to perform differential display and construction of cDNA libraries from the small amount of embryogenic tissue and outline a PCR-based method for confirming differential expression of large number of cloned bands in cases where RNA quantities are limiting.
PDF Abstract XML References Citation
How to cite this article
Ravindra B. Malabadi and K. Nataraja, 2007. Isolation of cDNA Clones of Genes Differentially Expressed During Somatic Embryogenesis of Pinus roxburghii. American Journal of Plant Physiology, 2: 333-343.
DOI: 10.3923/ajpp.2007.333.343
URL: https://scialert.net/abstract/?doi=ajpp.2007.333.343
DOI: 10.3923/ajpp.2007.333.343
URL: https://scialert.net/abstract/?doi=ajpp.2007.333.343
INTRODUCTION
Somatic embryogenesis has become the in vitro model for studying gene expression during early embryo development because of the difficulty in isolating early-stage zygotic embryos in vivo (Goldberg et al., 1994; Bishop-Hurley et al., 2003). Differential gene expression in somatic cells is involved in developmental program switching and confers on the somatic cells, the ability to manifest embryogenic potential. However, very few of the molecular events involved in the transition of a somatic cell to an embryogenic competent cell are known thus far (Xu et al., 1997; Bishop-Hurley et al., 2003; Stasolla et al., 2003; Goncalves et al., 2005a, b; Lorenz et al., 2005; Mathieu et al., 2006; Pullman et al., 2003). Various model systems have been widely investigated in order to understand the mechanisms of gene regulation during somatic embryogenesis using mature and immature zygotic embryos as the starting explants for the establishment of embryogenic culture system. The first evidence of differential gene expression during this developmental process was reported in the 1990s (Dong and Dunstan, 1996a) and to date, some genes that are activated or differentially expressed during the induction and development of somatic embryos have been cloned and studied by the use of various molecular techniques (Dong and Dunstan, 1996b, 1997, 1999). Nonetheless, most studies have concentrated on the later stages of embryogenesis, although most vital genes are expressed at high levels at the time of somatic embryogenic-culture-initiatation. No reports of gene expression studies for somatic embryogenesis using vegetative shoot apices or secondary needles of mature trees of pines are available in the literature, although somatic embryogenesis protocols using explants (shoot apex and secondary needles) of mature trees of different pines were recorded and well established (Malabadi and van Staden, 2003, 2006, 2005a-d; Malabadi et al., 2004; Malabadi and Nataraja, 2006a, 2007a, b).
The comprehensive and integrated mechanisms controlling plant gene expression during somatic embryogenesis using vegetatative shoot apices of mature trees of pines remain unreported. We believe that the identification and isolation of the vital genes of somatic embryogenesis and elucidation of the transcript expression patterns is a prerequisite for understanding the precise mechanisms controlling the differentiation of somatic cells and the detailed steps by which these genes direct somatic embryogenesis. It ultimately will provide deeper insight into understanding the enigmatic reprogramming of cells in higher plants and well surely provide novel target genes for the improvement of somatic embryogenesis ability, as well as other aspects of genetic engineering. The aim of our investigation was to isolate genes expressed due to cold-pretreatment of explants during initiation of embryogenic tissue derived from vegetative shoot apices of mature trees of Pinus roxburghii, for which no previous published reports are available in literature. This will certainly help in understanding cold-enhanced somatic embryogenesis of pines including other higher plants.
MATERIALS AND METHODS
Origin of Embryogenic Tissue: The embryogenic tissue was initiated on DCR (Gupta and Durzan, 1985) medium supplemented with 22.62 μM 2, 4-D, 26.85 μM NAA and 8.87 μM BA (initiation medium) according to our existing previous protocols (Malabadi et al., 2004; Malabadi and Nataraja, 2006a, b; Malabadi and van Staden, 2006). For the following experiments of gene cloning, two types of plant material, 1) embryogenic tissue initiated by cold-pre treatment. 2) control (non-embryogenic tissue induced without cold pre-treatment) have been used for the isolation of total RNA.
Isolation of Total RNA
Total RNA was isolated according to the protocol of Salzman et al. (1999) with slight modifications. The integrity of RNA sample was verified by examining the integrity of 28S and 18S ribosomal fragments after separating 15 μg of total RNA on 1.2% agarose gel containing 2.9% formaldehyde following denaturation of samples at 100°C for 2 min in formaldehyde and formamide.
Separation of Poly (A) + RNA
Poly (A) + RNA was seperated from total RNA using Oligo (dT)25 magnetic beads (Dynal, Oslo, Norway) following the manufacturers instructions with slight modifications. First pass poly (A)+ RNA was pooled and passed over the beads a second time before use as a template for cDNA synthesis. Poly (A) + RNA from this second purification was precipitated with 2x vol of ethanol and 0.1x vol of 3.0 M aqueous sodium acetate (pH 4.8), washed with 70% (v/v) aqueous ethanol and resuspended in LoTE buffer (3 M Tris-HCl, 0.2 mM EDTA, pH 7.5 at 25°C) to a final concentration of about 1 μg L-1 (Lorenz et al., 2005).
Differential Display of mRNA
Differential display was performed, based on the original method of Liang and Pardee (1992) with slight modifications according to Stratagene protocols (Stratagene, La Jolly, CA, USA). Here total RNA was isolated from cold-enhanced embryogenic tissue and from non-embryogenic tissue (control). A total of 2.5 μL (5 μg) of freshly diluted poly (A) + RNA were reverse transcribed in a 50 μL reaction volume containing 5 μL of 10x first-strand buffer, 3.0 μL of 10 mM dNTPs, 2 μL of 1.4 μg μL-1 of Linker-primer, 35.0 μL of DEPC-treated water, 1 μL of RNAse Block Ribonuclease inhibitor (40 U μL-1). Finally 1.5 μL of Strata Script RT (50 U μL-1) was added to the above reaction. The contents of the samples were gently spin downed in a microcentrifuge. Reverse transcription was performed by incubation at 42°C for 1 h and stopped by inactivating the Strata Script MMLV reverse transcriptase at 75°C for 5 min. The products of reverse transcription were stored at -20°C for recent use or at -70°C for longer use.
The amplification of cDNA samples was conducted in 20 μL reaction volumes. PCR was performed in the presence of the same linker primer as was used in the corresponding reverse transcription reaction, as well as a single arbitoary primers (P1, P2, P3, P4, P5, P6, P7, P8, P9 and P10) (Clontech Lab, Inc., Palo Alto, CA, USA) as an upstream primer. The PCR reaction mixture contained 1.5 μL cDNA template, 2.0 μL of 1.4 μg μL-1 of Linker-primer, 2.0 μL of 0.5 μM arbitory primer, 3.0 μL of 10 mM dNTPs, 2.5 U Taq DNA-Polymerase (Roche) and 1x PCR buffer. Reactions were made up to volume using DEPC-treated water and were overlaid with mineral oil before PCR to prevent evaporation. Amplification was carried out for 40 cycles at 95°C for 30 sec, 42°C for 20 sec and 72°C for 30 sec, followed by an additional extension period of 5 min at 72°C. Control reactions were included for each of the RNA isolates in which 1 μL of non-reverse transcribed RNA was used in the reactions with each of the above primers and arbitary primer (Clontech Lab, Inc., Palo Alto, CA, USA) in order to ensure that any amplification products detected were not due to amplification of contaminating genomic DNA.
After amplification, the samples were separated on a 6% polyacrylamide (19: 1 acrylamide: bisacrylamide)/ 8 M urea sequencing gel in 0.5x TAE buffer. Immediately before loading, 2 μL loading buffer (95% v/v formamide; 20 mM EDTA; 0.05% w/v bromophenol blue; 0.05 % w/v xylene cyanol FF) were added to 4 μL aliquots of the amplified cDNAs and the samples denatured at 75°C for 2 min. Gels were run at 70 W (constant power) for 2.5 h or until the xylene cyanol dye has migrated through the entire gel. The gels were dried and transferred on Whatmans 3 MM chromatography paper without fixing and exposed using Hyperfilm-βmax (Amersham Biosciences) autoradiography film for 5 days.
Following development of the film, gel slices containing the differentially expressed bands were excised and placed in 0.5 mL PCR tubes containing 100 μL DEPC-treated water. The samples were incubated for 10 min at room temperature and then boiled for 10 min at 99°C. The tubes were overlaid with 2-3 drops of mineral oil to prevent evaporation. The tubes were centrifuged for 5 min at maximum speed in a desktop microfuge and the supernatant liquid was transferred to 0.5 mL PCR tubes. Samples were stored at -70°C until further use. For reamplification of the cDNA isolated from the gel, 4 μL of the elute was PCR amplified in the presence of the same primers as were originally used during differential display. The reamplified fragments were electrophoresed in 1% agarose gels stained with ethidium bromide and visualized under UV light. Following reamplification, cDNA fragments were purified from the agarose gel with a QIAquick Gel Extraction Kit (Qiagen Inc., Valencia, CA) and cloned into pBluescript II vector (Stratagene, La Jolly, CA, USA). Transformation of XL10-Gold strain cells was then carried out according to the vector manufactures instructions.
Synthesis of cDNA
The cDNA was synthesized following the protocols contained in a cDNA synthesis Kit (Stratagene, La Jolla, CA, USA). All libraries were constructed using 5 μg of second-pass poly (A) + RNA as input for first-strand cDNA synthesis. The reaction was primed using an oligo-dT primer tailed with the recognition sequence for the XhoI restriction endonuclease. After second strand synthesis, EcoRI adapters were ligated to the 5’-end of the cDNA. The cDNAs were then double-digested with XhoI and EcoRI. The digested cDNAs were size-fractionated by electrophoresis through 1% agarose gels in 10 mM Tris-HCl, 5 mM sodium acetate, 0.5 mM EDTA, pH 7.8 buffer at 25°C. The cDNA ranging in size from 900 bp to 10 kb was purified from the agarose gel with a Q I A quick Gel Extraction Kit (Qiagen Inc., Valencia, CA). The gel-purified cDNA was quantified by UV absorbance and concentrated by precipitation with 2.5x vol of absolute ethanol, 0.5x vol of 7.5 M ammonium acetate and 5 μL of 5 mg mL-1 glycogen (Roche biochemicals, Gmbh, Germany). The cDNA was washed twice with 70% (v/v) aqueous ethanol and dissolved in LoTE buffer (3 M Tris-HCl, 0.2 mM EDTA, pH 7.5) at a final concentration of 0.25 μg μL-1.
cDNA Library Construction
The plasmid vector for library construction, pBluescript II vector (Stratagene, La Jolla, CA, USA), was digested to completion with EcoRI and XhoI, followed by treatment with calf intestinal alkaline phosphatase. Ligation reactions typically contained 0.5-1.5 μg of cDNA, 25-50 ng μL-1 of the pBluescript II vector DNA, 0.5 μL of 10x ligase buffer, 4 units μL-1 of high concentration T4 DNA ligase in 10.0 μL reaction volume. Ligation reactions were incubated overnight at 16°C. Fully ligated samples were extracted with phenol: chloroform (24: 1 v/v), precipitated as described above and resuspended in LoTE buffer (3 M Tris-HCl, 0.2 mM EDTA, pH 7.5). These concentrated plasmid libraries were used in 1-3 μL aliquots to transform XL10-Gold ultracompetent cells (Stratagene, La Jolla, CA, USA) with the ligation reactions. XL10-Gold is a McrA– and McrB– strain and does not restrict methylated DNA (Stratagene, La Jolla, CA, USA). Transformation was done according to the existing protocols of Stratagene (La Jolla, CA, USA). Colony forming units (cfu) were determined after plating 5-25 μL of a 10-1 dilution of transformed cells on LB agar plates supplemented with 100 μg mL-1 ampicillin.
The color selection was seen when plating on LB agar plates containing 100 μg mL-1 ampicillin, 80 μg mL-1 of fresh X-gal and 20 mM IPTG. Colonies containing vectors without inserts will be blue after incubation for 12-18 h at 37°C. Colonies with vectors containing inserts will remain white. Further enhancement of the blue color was obtained by placing plates at 4°C for 2 h following overnight growth at 37°C.
Amplification of pBluescript cDNA Library
The primary cDNA library was now being plated and screened. However, amplification of the primary cDNA library is desirable to produce a large and stable quantity of the library. This was achieved following manufacturer guidelines of Stratagene (La Jolla, CA, USA) by amplifying the plasmid libraries in 500 mL bottles of 2x LB agarose supplemented with 100 μg mL-1 ampicillin, using the semi-solid amplification method. Finally 6 serial dilutions were performed with 100 μL of the amplified library diluted in 900 μL of LB medium. 10 μL of the 10-5 and 10-6 dilutions were plated onto selection plates. Amplification of a primary library containing 1x106 total transformants was resulted in a stable library of at least 1x109 total transformants. Remaining amplified library was stored at -80°C until further analyses.
Screening of cDNA Library by PCR
cDNA libraries were screened using M13 primers (Clontech laboratories, Inc., USA) to determine the percentage of recombinant clones. This was done by using two gene-specific primers for verifying the target gene in the clone. During screening, 15 isolated colonies were picked up with sterile toothpicks and inoculated each into 50 μL of TE buffer in a separate 1.5 mL microcentrifuge tubes. Tubes were boiled for 5 min and a 25 μL PCR reaction was set up for the screening. PCR reaction mixture (Clontech laboratories, Inc, USA) consisting of 2.5 μL of 10x PCR buffer, 0.5 μL of 10 mM each of dNTP mixture, 0.5 μL of 20 μM sense primer, 0.5 μL of 20 μM anti sense primer, 19.5 μL of water and 0.5 μL of Taq DNA polymerase). Finally to this reaction 1.0 μL of boiled colony lysate was added. The PCR contents were mixed well and all the samples were overlaid with an equal volume of paraffin oil prior to undergoing following amplification cycles (Hybaid Thermal Reactor, Hybaid Ltd., England). The PCR was initiated with a denaturation step of 94°C for 30 sec at the beginning of the cycling regime. This was then followed by 25 cycles each comprising of 94°C denaturing temperature (30 sec), a 68°C annealing step (2 min) and a one cycle of 68°C extension step (5 min). After electrophoresis, 5 μL of PCR product was electrophoresed on a 1.2% TAE/agarose gel with DNA size markers at 70 V for approximately 5 h. Typical bands of 700 bp in size were seen for clones containing inserts and gels were used for following Southern blot hybridization.
Southern Blot Hybridization
Southern blot analyses were performed to verify the screening of the cDNA libraries. The gels were depurinated, denaturated, neutralized and fragmented plasmid DNA was transferred to a nylon membrane (Roche Diagnostics GmbH, Mannheim, Germany) by capillary transfer. Double stranded probe for cDNA inserts (700 bp or 0.7 kb) was labeled with digoxigenin-11-dUTP in the PCR conditions following the protocols of Roche biochemicals. For Southern blot hybridization of cDNA, the above anchored and arbitory primers were used (Clontech laboratories, Inc., USA).
In the secondary screening procedure, plasmid DNA was isolated from recombinant colonies using the alkaline lysis method of Felliciello and Chinali, (1993) and quantified spectrophotometrically. Plasmid DNA was purified from selected clones, digested with EcoRI and XhoI to release the cDNA inserts from the plasmid and electrophoretically separated by agarose electrophoresis. This DNA was again transferred to nylon membranes using the downword capillary transfer method as described above. Again inserts showing a signal for the induction of embryogenic tissue by cold pre-treatment cDNA and no signals for the non-embryogenic tissue were analyzed further.
Reverse-Northern Blot Hybridization
Reverse-northern analysis was performed according to a method modified from Mou et al. (1994) to confirm that the inserts were cold-enhanced embryogenic-tissue specific. Duplicate positively charged nylon membranes were cut to size and wet for 5 min in 0.4 M Tris-HCl (pH 7.5). Ten micrograms of each plasmid to be blotted was placed into a sterile microfuge tube. To this added 100 μL denaturing solution (0.25 N NaOH, 1 M NaCl) was added. The plasmid DNA was then allowed to denature for 10 min at room temperature. Fifty microliters of the denatured plasmid solution was loaded into each of the corresponding wells of the manifold over the duplicate membranes. The negative vector sequence was also included as a negative control on both membranes. Once all wells had been loaded, the samples were left for 30 min before vacuum was applied. The membranes were neutralized by placing them onto filter paper soaked with 0.5 M NaCl/0.5 M Tris-HCl (pH 7.5). After 5 min, the membranes were air dried and cross linked using UV illumination.
For the synthesis of probe for reverse-northern-analysis, ten μg of total RNA was used. First strand cDNA was prepared from total RNA isolated from embryogenic tissue specific and control using the method as described above. The RNase inhibitor was not included in the reaction and anchored primer was replaced with oligo d(T) primer. All other reagents described previously. The reaction was incubated at 37°C for 60 min. The reverse transcriptase was then denatured by heating the reactions to 95°C for 5 min, where after 2.5 μg of random primer mixture was added to the assays. The reactions were heated at 90°C for 10 min and then held on ice for 5 min. A 50 μL cocktail mixture for random priming was prepared as described above. Reactions were terminated by adding 1 μL of 0.5 M EDTA (pH 8.0). Each reaction was made up to 100 μL with TE buffer and unincorporated nucleotids were removed by column chromatograpy using Roche Sephadex G50 mini Quick Spin columns. The elute from the spin columns was then used directly as the probe.
The membranes were pre-washed in hybridization bottles in a HYBAID mini hybridization oven. The chemiluminescence’s detection of hybridization products was performed according to manufacture’s (Roche biochemical’s) instructions.
RESULTS AND DISCUSSION
In the present study of differential display gene expression, two different treatment conditions were selected to isolate genes which are differentially expressed during induction of embryogenic tissue from vegetative shoot apices of mature trees of Pinus roxburghii. The resulted embryogenic tissue due to cold-pretreatment was subcultured on maintenance medium and produced somatic embryos on maturation medium. Somatic embryos after successful germination produced plantlets according to our previous protocols (Malabadi and Nataraja, 2006a, b). On the other hand for the control treatments, non-embryogenic tissue failed to produce any cleavage polyembryony and somatic embryos on the maturation medium.
During the isolation of RNA samples, it was consistently observed that approximately four times the amount of RNA was isolated from cold-enhanced embryogenic tissue than was isolated from the control samples. Cold-enhanced embryogenic tissue yielded an average (mean) of 81.0± 9.1 μg RNA per gram fresh wt embryogenic tissue, as compared with 23.4± 6.2 μg RNA/g fresh wt of the control group (Fig. 1). The measurement of steady-state mRNA levels by RNA blot analyses is a useful approach to studies of differential gene expression. The isolation of intact total RNA can be a difficult and tedious step in this analysis. A conventional method for RNA isolation has several limitations. Isolation of RNA from plant tissues is further complicated by high levels of ribonuclease activity and by complex cell wall components which are solubilized upon extraction. Molecular investigation of many interesting phenomena in plants has been prevented by difficulties in extracting high-quality RNA from tissues containing high levels of phenolic compounds, carbohydrates and other unidentified compounds. Phenolic compounds are readily oxidized to form covalently linked quinines and avidly bind nucleic acids. This renders RNA unable for such fundamental procedures as reverse transcription and cDNA library construction. In the method presented here, phenolic compounds are bound to soluble PVP and then eliminated by ethanol precipitation of the RNA. Proteins and carbohydrates are subsequently removed by phenol extraction and LiCl precipitation respectively. In our present investigation, the RNA purity estimated spectrophotometrically gave ratio for OD. 260/280 of 1.8 to 1.95 and more than 2.0 for 260/230, indicating that there was no significant contamination with proteins and polysaccharides in the RNA extracts. When cultured tissues such as embryogenic cultured cells, calli or somatic embryos are used as source of RNA extraction, increased polysaccharide content has to be considered in the RNA extraction strategy. In this protocol, polysaccharides were removed by the addition of sodium acetate while washing the crude RNA pellets.
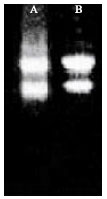 | |
| Fig. 1: | Agarose-formaldehyde denaturing gel electrophoresis of RNA. Total RNA was extracted from cold enhanced embryogenic tissue and control (non-embryogenic tissue) derived from vegetative shoot apices of mature trees of Pinus roxburghii. Lanes A (control, non-embryogenic tissue), B (cold enhanced embryogenic tissue) represents total RNA (28S, 18S distinct bands) |
This step has improved the final yield of good quality of RNA. Further as described above, this isolation procedure has the advantage of disrupting whole tissue samples rapidly and completely, while simultaneously inactivating RNase, even when it is present in greater abundance. The nuclear and organelle disruption that accompany membrane solubilization liberates nuclear RNA and genomic DNA, both of which co-purify with cytoplasmic RNA. The efficiency of protein denaturation (including disruption of RNAses) may be enhanced by the inclusion of â-mercaptoethanol, a reducing agent which breaks intra-molecular protein disulfide bonds. Incorporation of denaturing solution prepared by mixing saturated phenol with chloroform are organic solvents that very efficiently denature and cause the precipitation of proteins (Maniatis et al., 1982; Wan and Wilkins, 1994; Lefebvre et al., 1980). Chloroform stabilizes the phenol, imparts a greater density to this organic extracting material, improves the efficiency of deproteininzation of the sample and facilitates removal of lipids from the RNA preparation. Large RNAs were efficiently precipitated with LiCl(Salzman et al., 1999; Maniatis et al., 1982; Wan and Wilkins, 1994; Lefebvre et al., 1980). The contaminated polysaccharides were removed by addition of one volume of ethanol. Then RNA (Fig. 1) was concentrated by precipitation with sodium chloride and ethanol and collected after centrifugation. Figure 1 shows the presence of two narrow bands due to the 28S and 18S RNA, indicating good quality of intact RNA from cold-enhanced embryogenic tissue.
Magnetic bead separation of RNA has been employed in mRNA isolation and the isolated mRNA has been used in differential display (Xu et al., 1997; Goncalves et al., 2005a, b; Lorenz et al., 2005). The Dynal Oligo (dT)25 beads (Dynal, Oslo, Norway) can bind to 2 μg mRNA mL-1. When a small amount of tissue is used, most of the Oligo (dT)25 is wasted and may interfere in the subsequent PCR reaction. In our experiments also we reduced the bead quantity from the manufacturer’s recommendation of 50 μL sample-1 to 15 μL sample-1, an amount that is barely visible in the tube during the isolation. If a large amount of tissue is used, the amount of Oligo (dT)25 beads (Dynal, Oslo, Norway) can be increased. This method relies on A-T base pairing. Short sequences of oligo-dT are covalently bound to the surface of the Dynabeads and will hybridize to the poly A tail of the mRNA, allowing simple and rapid magnetic isolation. This allows the detection of mRNA by RT-PCR from highly specialized cells. High quality, full length mRNA is necessary for meaningful results in any gene expression study. Therefore, choosing the optimal mRNA sample preparation method is essential. Magnetic separation with Dynabeads Oligo (dT)25 is a rapid method to obtain pure, intact mRNA without any DNA or rRNA contamination.
The construction of cDNA libraries is fundamental in discovering new genes and assigning gene function. cDNA libraries constructed directly into plasmid vectors are convenient for plasmid-based functional screening, Expressed Sequence Tagged (EST) sequencing, normalization and substraction techniques. The cDNA was synthesized following the protocols contained in a cDNA synthesis Kit (Stratagene, La Jolla, CA, USA). All libraries were constructed using 5 μg of second-pass poly (A) + RNA as input for first-strand cDNA synthesis. The reaction was primed using an oligo-dT primer tailed with the recognition sequence for the XhoI restriction endonuclease. After second strand synthesis, EcoRI adapters were ligated to the 5’-end of the cDNA. The cDNAs were then double-digested with XhoI and EcoRI.
During the synthesis of cDNA, messenger RNA is primed in the first-strand synthesis with the linker-primer and is reverse-transcribed using StrataScriptTM reverse transcriptase (StrataScript RT) and 5-methyl dCTP. StrataScript reverse transcriptase is a novel Moloney murine leukemia virus reverse transcriptase (MMLV-RT) without any detectable RNase H activity. Cloned StrataScript RT is purified from recombinant E-coli containing a genetically engineered mutant MMLV-RT gene. A point mutation in the highly conserved residue of the RNase H region results in the loss of undesired RNase H degradative activity without affecting the desired reverse transcriptase function. The result is a nuclease-free mutant of MMLV-RT that can produce larger yields of full length cDNA transcripts than wild type MMLV-RT, which possesses substantial RNase H activity. The use of 5-methyl dCTP during first-strand synthesis hemimethylates the cDNA, which protects the cDNA from digestion with cetain restriction endonucleases such as XhoI. Therefore, on XhoI digestion of the cDNA, only unmethylated site within the linker-primer is cleaved.
The digested cDNAs were size-fractionated by electrophoresis through 1% agarose gels in 10 mM Tris-HCl, 5 mM sodium acetate, 0.5 mM EDTA, pH 7.8 buffer at 25°C. The cDNA ranging in size from 900 bp to 10 kb was purified from the agarose gel with a QIAquick Gel Extraction Kit (Qiagen Inc., Valencia, CA). Further cDNA was cloned using pBluescript vector, which is a high-copy-number pUC-based plasmid with ampicillin resistance and the convenience of the blue-white color selection. This vector is driven by the lac promoter, which is represented in the presence of the LacI protein and is inducible by isopropyl-â-D-thio-galactopyranoside (IPTG). In bacteria expressing the lacZÄM15 mutation and lacI, such as XL10-Gold cells, colonies containing vector without insert will be blue in the presence of 5-bromo-4-chloro-3-indoyl-â–D-galactopyranoside (X-gal) and IPTG. Ampicillin-resistant colonies containing vector with insert will be white and can express the promoter sequences are present in the N-terminal portion of a lacZ gene fragment.
Differential display was conducted using 40 different primer combinations (one linker-primer as was used in reverse transcription and 10 arbitory primers, P1-P10). Typically, approximately 65 cDNA bands ranging in size from 100 bp to over 1000 bp were resolved on the acrylamide gels per primer combination. Duplicate samples were thus run in adjacent lanes. The overall number of such fragments was very low. Such bands were easily distinguished and were disregarded during the analysis of the gels. Fifty-six cold-enhanced embryogenic tissue specific fragments (2% of the total number of cDNA bands generated from total RNA isolated from cold pretreatment induced embryogenic tissue) were used for the identification along with 110 cDNA fragments specific to the somatic embryogenesis (4% of the total). All the differentially expressed cDNA fragments were excised from the gels and the DNA eluted and re-amplified.
Amplification of the primary cDNA library is desirable to produce a large and stable quantity of the library (Fig. 2). This was achieved according to the available instructions of Stratagene (La Jolla, CA, USA) by amplifying the plasmid libraries in 500 mL bottles of 2x LB agarose supplemented with 100 μg mL-1 ampicillin, using the semi-solid amplification method.
 | |
| Fig. 2: | Screening of cDNA library by Southern hybridization. After electrophoresis, 5 μL of PCR product was electrophoresed on a 1.2% TAE/agarose gel. Typical bands of 700 bp in size were seen for clones containing inserts and gels were used for Southern blot hybridization. The chemiluminescence detection of hybridization products was performed according to manufacture’s (Roche biochemicals, Germany) instructions |
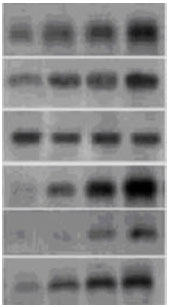 | |
| Fig. 3: | Reverse-Northern blot hybridization analysis of the cold enhanced somatic embryogenesis cDNAs representative of different types of gene expression patterns. 20 out of 24 clones have insert fragment. For hybridization, a fresh Easy Hyb Solution containing denatured cDNAs specific for cold-enhanced embryogenic tissue was used as a probe. This figure summarizes the results of several Northern hybridization experiments confirming the clones have insert fragments |
cDNA libraries were screened using M13 primers (Clontech laboratories, Inc., USA) to determine the percentage of recombinant clones. This was done by using two gene-specific primers for verifying the target gene in the clone. Southern blot analyses were performed to verify the screening of the cDNA libraries. In the secondary screening procedure, plasmid DNA was purified from selected clones, digested with EcoRI and XhoI to release the cDNA inserts from the plasmid and electrophoretically separated by agarose gel electrophoresis. This DNA was again transferred to nylon membranes using the downward capillary transfer method as described above. Again inserts showing a signal for the induction of embryogenic tissue by cold pre-treatment cDNA and no signals for the non-embryogenic tissue were analyzed further. Of the 56 cold-enhanced embryogenic tissue specific fragments, 20 could be re-amplified to produce a single band of the correct size on agarose gels (Fig. 3). Nine produced two bands, whilst a further 11 generated multiple bands. Thirty six cDNAs could not be re-amplified. Nine of the 20 fragments which generated single bands on re-amplification were selected for cloning and further analysis. During reverse northern hybridization, all the 20 clones selected generated a positive signal when probed with labeled cDNA from cold-enhanced embryogenic tissue, but no signal when probed with cDNA from the non-embryogenic tissue (control treatment). All the 20 clones thus contained inserts that were specific to cold-enhanced somatic embryogenesis (Fig. 3).
In conclusion, the package of procedures we have presented offers a fast, sensitive and reliable approach to the isolation and study of the expression of many genes that are differentially expressed during induction of embryogenic tissue in P. roxburghii. This work presents only preliminary experiments of gene expression. Our preliminary studies identified several differentially expressed mRNAs which might be homologous to regulatory proteins. The mRNA micro-preparation was effective when the RNA samples contained interfering material that otherwise rendered them poor templates for the RT-PCR reaction. In addition, little starting material was necessary for RT-PCR reactions; however, we think our procedure could be scaled up to prepare larger quantities of cDNA for many purposes. Present study has shown that somatic embryogenesis follows a unique developmental pathway regulated by temporal and spatial patterns of gene expression. Further studies of sequencing and searching through the databases, clustering and blasting are under study and will be presented in detail very soon. This will certainly highlights the identification and characterization of genes in programming the somatic cells towards effective somatic embryogenesis. This set of cold enhanced somatic embryo-specific genes is an important resource for understanding the genetic interactions underlying somatic embryogenesis signaling and regulation responses and may contribute to the characterization of the process of somatic embryogenesis in plants.
ACKNOWLEDGMENTS
The Head, Department of Botany, Karnatak University, Dharwad is warmly acknowledged for giving all the facilities for this research. Help rendered by the friends during this investigation is very much appreciated.
REFERENCES
- Bishop-Hurley, S.L., R.C. Gardner and C. Walter, 2003. Isolation and molecular characterization of genes expressed during somatic embryo development in Pinus radiata. Plant Cell Tissue Org. Cult., 74: 267-281.
CrossRef - Dong, J.Z. and D.I. Dunstan, 1996. Characterization of three heat-shock-protein genes and their developmental regulation during somatic embryogenesis in white spruce (Picea glauca). Planta, 200: 85-91.
Direct Link - Dong, J.Z. and D.I. Dunstan, 1996. Expression of abundant mRNAs during somatic embryogenesis of white spruce (Picea glauca). Planta, 199: 459-466.
Direct Link - Dong, J.Z. and D.I. Dunstan, 1997. Endochitinase and beta-1, 3-glucanase genes are developmentally regulated during somatic embryogenesis in Picea glauca. Planta, 201: 189-194.
Direct Link - Dong, J.Z. and D.I. Dunstan, 1999. Cloning and characterization of six embryogenesis-associated cDNAs from somatic embryos of Picea glauca and their comparative expression during zygotic embryogenesis. Plant Mol. Biol., 39: 859-864.
Direct Link - Feliciello, I. and G. Chinali, 1993. A modified alkaline lysis method for the preparation of highly purified plasmid DNA from Escherichia coli. Anal. Biochem., 212: 394-401.
Direct Link - Goldberg, R.B., G. De-Paiva and R. Yadegari, 1994. Plant embryogenesis: Zygote to seed. Science, 266: 605-614.
CrossRefDirect Link - Goncalves, S., J. Cairney, M. Oliveira and C. Miguel, 2005. Identification of differentially expressed genes during embryogenesis in Maritime pine (Pinus pinaster). Silva Lus., 13: 203-216.
Direct Link - Gupta, P.K. and D.J. Durzan, 1985. Shoot multiplication from mature trees of douglas fir and sugar pine. Plant Cell Rep., 4: 177-179.
CrossRef - Liang, P. and A.B. Pardee, 1992. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science, 257: 967-971.
CrossRefPubMedDirect Link - Lorenz, W.W., F. Sun, C. Liang, D. Kolychev and H. Wang et al., 2005. Water stress-responsive genes in loblolly pine (Pinus taeda) roots identified by analyses of expressed sequence tag libraries. Tree Physiol., 26: 1-16.
Direct Link - Malabadi, R.B. and J. Van Staden, 2003. Somatic embryos can be induced from shoot apical domes of mature Pinus patula trees. S. Afr. J. Bot., 69: 450-451.
Direct Link - Malabadi, R.B., H. Choudhury and P. Tandon, 2004. Initiation, maintenance and maturation of somatic embryo from thin apical dome section in Pinus kesiya (Royle ex. Gord) promoted by partial desiccation and Gellan gum. Sci. Hortic., 102: 449-459.
Direct Link - Malabadi, R.B. and J. Van Staden, 2005. Somatic embryogenesis from vegetative shoot apices of mature trees of Pinus patula. Tree Physiol., 25: 11-16.
Direct Link - Malabadi, R.B. and J. Van-Staden, 2005. Role of antioxidants and amino acids on somatic embryogenesis of Pinus patula. In vitro Cell Dev. Biol. Plant, 41: 181-186.
Direct Link - Malabadi, R.B. and J. Van-Staden, 2005. Storability and germination of sodium alginate encapsulated somatic embryos derived from the vegetative shoot apices of mature Pinus patula trees. Plant Cell Tiss. Org. Cult., 82: 259-265.
CrossRefDirect Link - Malabadi, R.B. and J. Staden-Van, 2006. Cold-enhanced somatic embryogenesis in Pinus patula is mediated by calcium. S. Afr. J. Bot., 72: 613-618.
CrossRefDirect Link - Malabadi, R.B. and K. Nataraja, 2006. Cryopreservation and plant regeneration via somatic embryogenesis using shoot apical domes of mature Pinus roxburghii Sarg. Trees. In Vitro Cell. Dev. Biol-Plant., 42: 152-159.
Direct Link - Malabadi, R.B. and K. Nataraja, 2006. RAPD detect no somaclonal variation in cryopreserved cultures of Pinus roxburghii SARG. Prop. Orn. Plants, 6: 114-120.
Direct Link - Malabadi, R.B. and K. Nataraja, 2007. Plant regeneration via somatic embryogenesis using secondary needles of mature trees of Pinus roxburghii sarg. Int. J. Bot., 3: 40-47.
CrossRefDirect Link - Maniatis, T., E.F. Fritsch and J. Sambrock, 1982. Molecular Cloning: A Laboratory Manual. 3rd Edn., Cold Spring Harbor Laboratory, Cold Spring Harbor, New York, USA.
Direct Link - Mathieu, M., M.A. Lelu-Walter, A.S. Blervacq, H. David, S. Hawkins and G. Neutelings, 2006. Germin-like genes are expressed during somatic embryogenesis and early development of conifers. Plant Mol. Biol., 61: 615-627.
Direct Link - Pullman, G.S., S. Johnson, G. Peter, J. Cairney and N. Xu, 2003. Improving loblolly pine somatic embryo maturation: Comparison of somatic and zygotic embryo morphology, germination and gene expression. Plant Cell Rep., 21: 747-758.
Direct Link - Salzman, R.N., T. Fujita, K. Zhu-Salzman, P.M. Hasegawa and R.A. Bressan, 1999. An improved RNA isolation method for plant tissues containing high levels of phenolic compounds or carbohydrates. Plant Mol. Biol. Rep., 17: 11-17.
Direct Link - Stasolla, C., L.V. Zyl, U. Egertsdotter, D. Craig, W. Liu and R.R. Sederoff, 2003. The effects of polyethylene glycol on gene expression of developing white spruce somatic embryos. Plant Physiol., 131: 49-60.
Direct Link - Wan, C.Y. and T.A. Wilkins, 1994. A modified hot borate method significantly enhances the yield of high quality RNA from cotton (Gossypium hirsutum L.). Anal. Biochem., 223: 7-12.
Direct Link - Xu, N., B. Johns, G. Pullman and J. Cairney, 1997. Rapid and reliable differential display from minute amounts of tissue: Mass cloning and characterization of differentially expressed genes from loblolly pine embryos. Plant Mol. Rep., 15: 377-391.
Direct Link