
V. Balamuralidhara
JSS College of Pharmacy, JSS University, Mysore, Karnataka-570015, India
LiveDNA: 91.37090
T.M. Pramodkumar
JSS College of Pharmacy, JSS University, Mysore, Karnataka-570015, India
N. Srujana
JSS College of Pharmacy, JSS University, Mysore, Karnataka-570015, India
M.P. Venkatesh
JSS College of Pharmacy, JSS University, Mysore, Karnataka-570015, India
N. Vishal Gupta
JSS College of Pharmacy, JSS University, Mysore, Karnataka-570015, India
K.L. Krishna
JSS College of Pharmacy, JSS University, Mysore, Karnataka-570015, India
H.V. Gangadharappa
JSS College of Pharmacy, JSS University, Mysore, Karnataka-570015, India
American Journal of Drug Discovery and Development
Year: 2011 | Volume: 1 | Issue: 1 | Page No.: 24-48







ABSTRACT
pH Sensitive Drug Delivery Systems (PSDDS) are gaining importance as these systems deliver the drug at specific time as per the pathophysiological need of the disease, resulting in improved patient therapeutic efficacy and compliance. Diseases wherein PSDDS are promising include asthma, peptic ulcer, cardiovascular diseases, cancer and hypertension. The specific time that patients take their medication is very important as it has significant impact on treatment success. Optimal clinical outcome cannot be achieved if drug plasma concentrations are constant. If symptoms of a disease display circadian variation, drug release should also vary over time. Drug pharmacokinetics can also be pH-sensitive; therefore, variations both in a disease state and in drug plasma concentration need to be taken into consideration in developing drug delivery systems intended for the treatment of disease with adequate dose. PSDDS wherein the drug release is controlled primarily by the delivery system, stimuli induced PSDDS in which release is controlled by the stimuli, such as the pH present in the intestinal tract. These systems are useful to several problems encountered during the development of a pharmaceutical dosage form. These, as well as pertinent issues, are addressed in this review.
PDF Abstract XML References Citation
Received: April 12, 2010;
Accepted: May 30, 2010;
Published: September 28, 2010
How to cite this article
V. Balamuralidhara, T.M. Pramodkumar, N. Srujana, M.P. Venkatesh, N. Vishal Gupta, K.L. Krishna and H.V. Gangadharappa, 2011. pH Sensitive Drug Delivery Systems: A Review. American Journal of Drug Discovery and Development, 1: 24-48.
DOI: 10.3923/ajdd.2011.24.48
URL: https://scialert.net/abstract/?doi=ajdd.2011.24.48
DOI: 10.3923/ajdd.2011.24.48
URL: https://scialert.net/abstract/?doi=ajdd.2011.24.48
INTRODUCTION
pH sensitive systems: Controlled drug delivery systems, which are intended to deliver drugs at predetermined rates for predefined periods of time, have been used to overcome the shortcomings of conventional drug formulations. Although, significant progress has been made in the controlled drug delivery area, more advances are yet to be made for treating many clinical disorders, such as diabetes and rhythmic heart disorders. In these cases, the drug has to be delivered in response to pH in the body. In fact, it would be most desirable if the drugs could be administered in a manner that pre- cisely matches physiological needs at proper times (temporal modulation) and/or at the proper site (site-specific targeting). In addition, the controlled drug delivery area needs further development of techniques for delivery of peptide and protein drugs. In the body, the appearance of numerous bioactive peptides is tightly controlled to maintain a normal metabolic balance via a feedback system called ‘homeostasis’ (Yoshida et al., 1993). It would be highly beneficial if the active agents were delivered by a system that sensed the signal caused by disease, judged the magnitude of signal and then acted to release the right amount of drug in response. Such a system would require coupling of the drug delivery rate with the physiological need by means of some feedback mechanism.
The pH range of fluids in various segments of the gastrointestinal tract may provide environmental stimuli for responsive drug release. Studies by several research groups (Brannon-Peppas and Peppas, 1989; Annaka and Tanaka, 1992; Firestone and Siegel, 1988; Dong and Hoffman, 1990a; Kou et al., 1990; Pradny and Kopecek, 1990; Siegel et al., 1988; Kono et al., 1993; Hariharan and Peppas, 1993; Siegel and Firestone, 1988; Allcock and Ambrosio, 1996; Bell and Peppas, 1996; Jarvinen et al., 1998) have been performed on polymers containing weakly acidic or basic groups in the polymeric backbone. The charge density of the polymers depends on pH and ionic composition of the outer solution (the solution into which the polymer is exposed). Altering the pH of the solution will cause swelling or deswelling of the polymer. Thus, drug release from devices made from these polymers will display release rates that are pH sensitive. Polyacidic polymers will be unswollen at low pH, because the acidic groups will be protonated and hence unionized. With increasing pH, Polyacidic polymers will swell. The opposite holds for polybasic polymers, because the ionization of the basic groups will increase with decreasing pH. Siegel et al. (1992) found the swelling properties of the polybasic gels are influenced also by buffer composition (concentration and pka). A practical consequence proposed is that these gels may not reliably mediate pH sensitive swelling controlled release in oral applications, because the levels of buffer acids in the stomach (where swelling and release are expected to occur) generally cannot be controlled. However, the gels may be useful as mediators of pH-triggered release when precise rate control is of secondary importance. This approach utilizes the existence of the pH gradient in the GIT that increases progressively from the stomach (pH 1.5-3.5) and small intestine (pH 5.5-6.8) to the colon (6.4-7.0). The most commonly used pH-sensitive polymers are derivatives of acrylic acid and cellulose. By combining the knowledge of polymers and their solubility at different pH environments, delivery systems have been designed to deliver drugs at the target site.
Annaka and Tannaka (1992), reported that more than two phases (swollen and collapsed) can be found in gels consisting of copolymers of randomly distributed positively and negatively charged groups. In these gels, polymer segments interact with each other through attractive or repulsive electrostatic interactions and through attractive or repulsive electrostatic interactions and through hydrogen bonding. The combination of these forces seems to in the existence of several phases, each characterized by a distinct degree of swelling, with abrupt jumps between them. The existence of these phases presumably reflects the ability of macromolecular systems to adopt different stable conformations in response to changes in environmental conditions. For copolymer gels prepared from acrylic acid (the anionic constituent) and methacryl-amido propyl-trimethyl ammonium chloride, the largest number of phases was seven. A similar approach was proposed by Bell and Peppas (1996); membranes made from grafted poly (methacrylic acid-g-ethylene glycol) copolymer showed pH sensitivity due to complex formation and dissociation. Uncomplexed equilibrium swelling ratios were 40 to 90 times higher than those of complexed states and varied according to copolymer composition and polyethylene glycol graft length.
Giannos et al. (1995) proposed temporally controlled drug delivery systems, coupling pH oscillators with membrane-diffusion properties. By changing the pH of a solution relative to the pka, a drug may be rendered charged or uncharged. Because only the uncharged form of a drug can permeate across lipophilic membranes, a temporally modulated delivery profile may be obtained with a pH oscillator in the donor solution.
Heller and Trescony (1979) were the first to propose the use of pH sensitive bioerodible polymers. In their approach, described in the section on systems utilizing enzymes, an enzyme-substrate reaction produces a pH change that is used to modulate the erosion of a pH sensitive polymer containing a dispersed therapeutic agent.
Bioerodible hydrogels containing azoaromatic moieties were synthesized by Ghandehari et al. (1997). Hydrogels with lower cross-linking density underwent a surface erosion process and degraded at a faster rate. Hydrogels with higher cross-linking densities degraded at a slower rate by a process where the degradation front moved inward to the center of the polymer.
Recently recombinant DNA methods were used to create artificial proteins that undergo reversible gelation in response to changes in pH or temperature (Petka et al., 1998). The proteins consist of terminal leucine zipper domains flanking a central, flexible, water soluble polyelectrolyte segment. Formation of coiled-coil aggregates of the terminal domains in near neutral aqueous solutions triggers formation of a three dimensional polymer network, with the polyelectrolyte segment retaining solvent and preventing precipitation of the chain. Dissociation of the coiled-coil aggregates through elevation of pH or temperature causes dissolution of the gel and a return to the viscous behavior that is characteristic of polymer solution. The authors suggest these hydrogels have potential in bioengineering applications requiring encapsulation or controlled release of molecules and cellular species.
Advantages: Drug directly available at the target site:
| • | Decreased dose to be administered |
| • | Decreased side effect |
| • | Improved drug utilization |
| • | Improved patient compliance |
| • | Lower daily cost to patient due to fewer dosage units are required by the patient in therapy |
| • | Drug adapts to suit circadian rhythms of body functions or diseases |
| • | Drug targeting to specific site like colon |
| • | Protection of mucosa from irritating drugs |
| • | Drug loss is prevented by extensive first pass metabolism |
DISEASES TARGETING pH SENSITIVE DRUG DELIVERY
Up to now, design of drug delivery systems has been governed by the homeostatic theory. This theory is based on the assumption of biological functions that display constancy over time. However, chronobiological studies have established circadian rhythm for almost all body functions, e.g., heart rate, blood pressure, body temperature, plasma concentration of various hormones, gastric pH and renal function (Li, 2003). It has become apparent that rhythmic processes are indispensable for the treatment of human diseases. Just as physiological functions vary over time, pathological states of disease have circadian rhythms. Epidemiological studies have documented the elevated risk of disease symptoms during the 24 h cycle (Fig. 1) (Smolensky and Labreque, 1997).
Thorough understanding of the disease physiology is required before designing the pH sensitive drug delivery system. Diseases where rhythmic circadian organization of the body plays an important role, pharmacokinetics and/or pharmacodynamics of the drugs is not constant within 24 h.
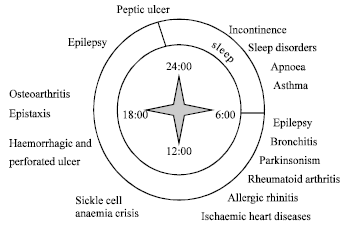 |
| Fig. 1: | Diseases lnown to display circadian rhythm |
Anti-ulcer therapy: It is well established that patients with peptic ulcer disease often experience the greatest degree of pain near the time that they go to bed, as the rate of stomach acid secretion is highest at night (Evans and Marain, 1996). The timing of administration of ulcer medications has a significant impact on their therapeutic effect.
Anti-inflammatory therapy: In the case of individuals who suffer from rheumatoid arthritis and related painful joint disorders, the non-steroidal anti-inflammatory agents (NSAIDs) such as ibuprofen may be more effective at relieving pain, if the drug is administered at least 4 to 6 h before the pain reaches its peak. It will be more helpful if arthritis patients take the NSAIDs before bed time if they experience a particularly high level of discomfort in the morning (Smolensky and Labreque, 1997).
Anti-asthma therapy: It has been estimated that symptoms of asthma occur 50 to 100 times more often at night than during the day (Washington and Wilson, 2009). Many circadian-sensitive factors appear to contribute to the worsening of nocturnal asthmatic symptoms. For example, cortisol (an anti-inflammatory substance) levels were highest at the time of awakening and lowest in the middle of the night and histamine (a mediator of bronchoconstriction) concentrations peaked at a level that coincided with the greatest degree of bronchoconstriction at 4:00 am (Ura et al., 1992). A research finding also reveals that theophylline absorption is slower at night (Lamberg, 1991). The enhanced understanding of the chronobiological impact upon the pathology of asthma and the pharmacology and pharmacokinetics of the drugs used in its management, have led to new approaches to disease management and enhanced patient care.
Cardiovascular therapy: The differences in patterns of illness between day and night for cardiovascular disorders such as hypertension, angina, heart attack, sudden cardiac death and stroke have been documented (Evans and Marain, 1996). Medications have been formulated and dosing schedules established, in an attempt to provide appropriate concentration of a drug in the target area of the body when the drug is most needed (Evans and Marain, 1996). For example, it has often been found that the blood pressure of a hypertensive patient increases rapidly in the morning after awakening, typically peaks in the middle to late time of the day, decreases in the evening and is lowest while the patient sleeps at night (Evans and Marain, 1996). It may also be important to recognize that the risk of heart attack appears to be greatest during the early morning hours after awakening. Currently, there are antihypertensive products in the market that are chronotherapeutic medications with novel drug delivery systems, releasing drug during the vulnerable period of 6 am to noon upon administration of medications at 10 pm.
Chemotherapy: Antineoplastic drugs cause cytotoxic effectson healthy and diseased tissues. As would be expected, the biological rhythms of both healthy and tumor cells may influence the susceptibility of normal and malignant cells to these agents (Washington and Wilson, 2009). It has been demonstrated that susceptibility rhythms to drugs may differ between healthy tissue and cancerous tissue. Therefore, the correct timing of drug treatment may reduce host toxicity, increase maximum drug tolerance and ultimately result in better tumor management. The pharmacologic and pharmacokinetic properties of the drug, rhythmic changes in DNA and RNA synthesis, RNA translational activity and mitotic activity may influence tumor cell susceptibility (Ura et al., 1992). It appears that the timing of drug administration in the treatment of cancer can have a significant impact upon treatment success.
Colonic drug therapy: The colon is also viewed as the preferred absorption site for oral administration of protein and peptide drugs, because of the relatively low proteolytic enzyme activities in the colon. In addition, to providing more effective therapy of colon related diseases such as irritable bowel syndrome, Inflammatory Bowel Disease (IBD) including Crohn’s disease and ulcerative colitis, colon specific delivery has the potential to address important unmet therapeutic needs including oral delivery of macromolecular drugs Therefore, it appears that targeted drug delivery with an appropriate release pattern could be crucial in providing effective therapy for this chronic disease (Yang et al., 2002).
METHODOLOGIES FOR pH SENSITIVE DRUG DELIVERY
pH-sensitive hydrogels
Polymer structures: All the pH-sensitive polymers contain pendant acidic (e.g. carboxylic and sulfonic acids) or basic (e.g., ammonium salts) groups that either accept or release protons in response to changes in environmental pH. The polymers with a large number of ionizable groups are known as polyelectrolytes. Figure 2 shows structures of examples of anionic and cationic polyelectrolytes and their pH-sensitive ionization. Poly (acrylic acid) (PAA) becomes ionized at high pH, while poly (N, N-diethylaminoethyl methaacrylate) (PDEAEM) becomes ionized at low pH. As shown in Fig. 2, cationic polyelectrolytes, such as PDEAEM, dissolve more, or swell more if cross linked, at low pH due to ionization. On the other hand, polyanions, such as PAA, dissolve more at high pH.
Properties of pH-sensitive hydrogels: Hydrogels made of crosslinked polyelectrolytes display big differences in swelling properties depending on the pH of the environment. The pendant acidic or basic groups on polyelectrolytes undergo ionization just like acidic or basic groups of monoacids or monobases. Ionization on polyelectrolytes, however, is more difficult due to electrostatic effects exerted by other adjacent ionized groups. This tends to make the apparent dissociation constant (K a) different from that of the corresponding monoacid or monobase. The presence of ionizable groups on polymer chains results in swelling of the hydrogels much beyond that can be achievable by nonelectrolyte polymer hydrogels. Since the swelling of polyelectrolyte hydrogels is mainly due to the electrostatic repulsion among charges present on the polymer chain, the extent of swelling is influenced by any condition that reduce electrostatic repulsion, such as pH, ionic strength and type of counter ions (Firestone and Siegel, 1991).
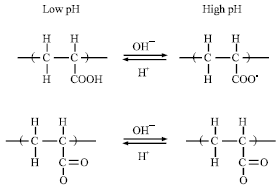 |
| Fig. 2: | pH-sensitive ionization of polyelectrolytes. Poly (acrylic acid) (top) and poly (N, N-diethylaminoethyl methacrylate) (bottom) |
The swelling and pH-responsiveness of poly- electrolyte hydrogels can be adjusted by using neutral comonomers, such as 2-hydroxyethyl methacrylate, methyl methacrylate and maleic anhydride (Falamarzian and Varshosaz, 1998; Kou et al., 1988; Brannon-Peppas and Peppas, 1990; Khare and Peppas, 1993). Different comonomers provide different hydrophobicity to the polymer chain, leading to different pH-sensitive behavior. Hydrogels made of poly (methacrylic acid) (PMA) grafted with poly (ethylene glycol) (PEG) have unique pH-sensitive properties (Peppas and Klier 1991). At low pH, the acidic protons of the carboxyl groups of PMA interact with the ether oxygen of PEG through hydrogen bonding and such complexation results in shrinkage of the hydrogels. As the carboxyl groups of PMA become ionized at high pH, the resulting decomplexation leads to swelling of the hydrogels. The same principle can be applied to IPN systems where two different types of polymer chain interact through pH-sensitive hydrogen bonding.
Applications of pH-sensitive hydrogels
Controlled drug delivery: pH-sensitive hydrogels have been most frequently used to develop controlled release formulations for oral administration. The pH in the stomach (<3) is quite different from the neutral pH in the intestine and such a difference is large enough to elicit pH sensitive behavior of polyelectrolyte hydrogels. For polycationic hydrogels, the swelling is minimal at neutral pH, thus minimizing drug release from the hydrogels. This property has been used to prevent release of foul-tasting drugs into the neutral pH environment of the mouth. When caffeine was loaded into hydrogels made of copolymers of methyl methacrylate and N, N-dimethylaminoethylmethacrylate (DMAEM), it was not released at neutral pH, but released at zero-order at pH 3-5 where DMAEM became ionized (Siegel et al., 1988). Polycationic hydrogels in the form of semi-IPN have also been used for drug delivery in the stomach. Semi-IPN of crosslinked chitosan and PEO showed more swelling under acidic conditions (as in the stomach). This type of hydrogels would be ideal for localized delivery of antibiotics, such as amoxicillin and metronidazole, in the stomach for the treatment of Helicobacter pylori (Patel and Amiji, 1996).
Hydrogels made of PAA or PMA can be used to develop formulations that release drugs in a neutral pH environment (Brannon-Peppas and Peppas, 1990; Khare and Peppas, 1993; Vishal Gupta et al., 2007). Hydrogels made of polyanions (e.g., PAA) crosslinked with azoaromatic crosslinkers were developed for colon-specific drug delivery. Swelling of such hydrogels in the stomach is minimal and thus, the drug release is also minimal.
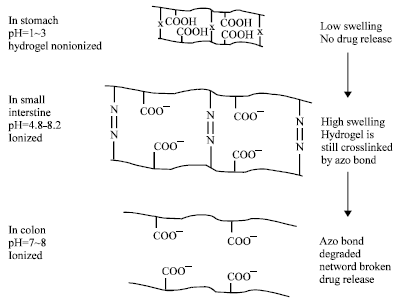 |
| Fig. 3: | Schematic illustration of oral colon-specific drug delivery using biodegradable and pH-sensitive hydrogels |
The extent of swelling increases as the hydrogel passes down the intestinal tract due to increase in pH leading to ionization of the carboxylic groups. But, only in the colon, can the azoaromatic cross-links of the hydrogels be degraded by azoreductase produced by the microbial flora of the colon (Ghandehari et al., 1997; Akala et al., 1998), as shown in Fig. 3. The degradation kinetics and degradation pattern (e.g., surface erosion or bulk erosion) can be controlled by the crosslinking density (Ghandehari et al., 1997). The kinetics of hydrogel swelling can be controlled by changing the polymer composition (Akala et al., 1998). The polymer composition can be changed as the pH of the environment changes. Some pendant groups, such as N-alkanoyl (e.g., propionyl, hexanoyl and lauroyl) and O-acylhydroxylamine moieties, can be hydro- lyzed as the pH changes from acidic to neutral values and the rate of side-chain hydrolysis is sensitive on the length of the alkyl moiety.
pH-sensitive hydrogels were placed inside capsules (Gutowska et al., 1997) or silicone matrices (Bilia et al., 1996; Carelli et al., 1999) to modulate the drug release. In the squeezing hydrogel system (Gutowska et al., 1997), drug release was controlled by a mechanism shown in Fig. 4. The only difference is that the swelling-shrinking of hydrogels is con- trolled by changing pH, instead of temperature. In the silicone matrix system (Bilia et al., 1996; Carelli et al., 1999), medicated pH-sensitive hydrogel particles made of semi-IPN of PAA and PEO were used. The release patterns of several model drugs having different aqueous solubilities and partitioning properties (including salicylamide, nicotinamide, clonidine HCl and prednisolone) were correlated with the pH-sensitive swelling pattern of the semi-IPN. At pH 1.2, the network swelling was low and the release was limited to an initial burst. At pH 6.8, the network became ionized and higher swelling resulted in increased release.
Superporous hydrogels (Vishal Gupta and Shivakumar, 2009) for delivery of drug in the alkaline pH were formulated employing acrylamide and methacrylic acid by free radical polymerization. These swelled only in basic pH and showed very fast swelling kinetics. Superporous hydrogels (Vishal Gupta and Shivakumar, 2010) have also been developed as gastoretentive drug delivery system as they swell only in acidic pH and are highly pH sensitive.
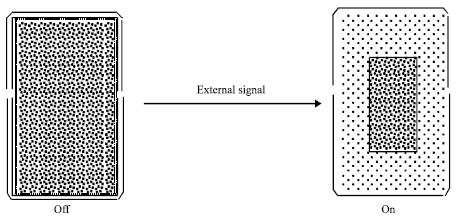 |
| Fig. 4: | Schematic illustration of on-off release from a squeezing hydrogel device for drug delivery |
Poly (vinylacetaldiethylaminoacetate) (PVD) has pH-sensitive aqueous solubility. Both the turbidity and SEM results showed that PVD formed a hydro-gel upon increase in pH from 4 to 7.4 (Aikawa et al., 1998a). The release of a model drug, chlorpheniramine maleate, was fast right after the PVD solution was introduced into a pH 7.4 buffer solution, but became very slow after the PVD hydrogel was formed (Aikawa et al., 1998a). The pH-sensitive sol-to-gel transformation of AEA was used to develop nasal spray dosage forms for treating allergic rhinitis and sinusitis (Aikawa et al., 1998b). The in vivo rat study showed that the apparent disappearance rate constant of chlorpheniramine maleate decreased with increase in the PVD concentration. The hydrogel formation on the mucous membranes in the rat nasal cavity was visually confirmed. If the time for sol-to-gel transition is shortened and the mucoadhesive property is added, the PVD system could be an ideal system for nasal delivery.
Hydrogels that are responsive to both temperature and pH can be made by simply incorporating ioniz-able and hydrophobic (inverse thermosensitive) functional groups to the same hydrogels. When a small amount of anionic monomer, such as acrylic acid, is incorporated in a thermoreversible polymer, the LCST of the hydrogel depends on the ionization of the pendant carboxyl groups, i.e., the pH of the medium. As the pH of the medium increases above the pK of the carboxyl groups of polyanions, LCST shifts to higher temperatures due to the increased hydrophilicity and charge repulsion. Terpolymer hydrogels made of NIPAAm, vinyl terminated poly-dimethylsiloxane macromer and acrylic acid was used for the delivery of indomethacin and amylase (Dong and Hoffman, 1990b, 1991). Other terpolymer hydrogels containing NIPAAm, acrylic acid and 2-hydroxyethyl methacrylate were prepared for the pulsatile delivery of streptokinase and heparin as a function of stepwise pH and temperature changes (Vakkalanka et al., 1997; Brazel and Peppas, 1996).
Other applications: pH-sensitive hydrogels have also been used in making biosensors and permeation switches (Hoffman, 1997). The pH-sensitive hydrogels for these applications are usually loaded with enzymes that change the pH of the local microenvironment inside the hydrogels. One of the common enzymes used in pH-sensitive hydrogels is glucose oxidase which transforms glucose to gluconic acid. The formation of gluconic acid lowers the local pH, thus affecting the swelling of pH-sensitive hydrogels.
Limitations and improvements: One of the inherent limitations of synthetic pH sensitive polymers is their non-biodegradability. For this reason, hydrogels made of non-biodegradable polymers have to be removed from the body after use. The non-biodegradability is not a problem in certain applications, such as in oral drug delivery, but it becomes a serious limitation in other applications, such as the development of implantable drug attention has been focused on the development of biodegradable, pH-sensitive hydrogels based on polypeptides, proteins and polysaccharides (Chiu et al., 1999; Markland et al., 1999). Dextran was activated with 4-aminobutyric acid for crosslinking with 1, 10-diaminodecane and also grafted with carboxylic groups (Chiu et al., 1999). The modified dextran hydrogels showed a faster and higher degree of swelling at high pH conditions and changing the pH between 7.4 and 2.0 resulted in cyclic swelling-deswelling. It is noted that dextran hydrogels may not be exactly biodegradable, since the body or certain sites in the body may not have the enzyme to degrade dextran molecules. Natural polysaccharides are not necessarily biodegradable in the human body.
Synthetic polypeptides were also used in synthesis of biodegradable hydrogels because of their more regular arrangement and less versatile amino acid residues than those derived from natural proteins. Examples of such synthetic polypeptide hydrogels include poly(hydroxyl-L-glutamate), poly(L-or-oxinithine), poly(aspartic acid), poly(L-lysine) and poly-(L-glutamic acid) (Markland et al., 1999). In addition to normal electro-static effects associated with most pH-sensitive synthetic polymer hydrogels, secondary structures of the polypeptide backbone may also contribute to the pH-sensitive swelling behavior (Markland et al., 1999). The overall extent of pH-responsive swelling could be en-gineered by modification of the polypeptide by changing its hydrophobicity and degree of ionization.
Enteric-coated systems: Enteric-coated formulations are suitable vehicles to modify the release of active substances such that release at specific target areas within the gastrointestinal (GI) tract can be affected, although the effectiveness of this methodology has long been a point of discussion. Kramer et al. (1996) investigated the use of enteric coatings of 261 pharmaceutical products (Fig. 5). The intended use included taste (9.6%) and odor (1%) masking, drug stabilization (31%), protection against local irritation (38%) and release directed to defined segments in the digestive tract (51%). Enteric coatings have traditionally been used to prevent the release of a drug in the stomach (Fig. 6). A major aim of enteric coating is protection of drugs that are sensitive or unstable at acidic pH. This is particularly important for drugs such as enzymes and proteins, because these macromolecules are rapidly hydrolyzed and inactivated in acidic medium. Antibiotics, especially macrolide antibiotics like erythromycin, are also rapidly degraded by gastric juices. Others, such as acidic drugs like NSAID’s (e.g., diclofenac, valproic acid, or acetylsalicylic acid) need to be enteric coated to prevent local irritation of the stomach mucosa.
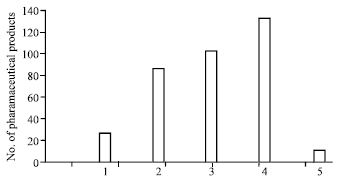 |
| Fig. 5: | Functions of enteric coatings according to the statements of the pharmaceutical manufacturer. 1, Taste masking; 2, stability; 3, protection against local irritation; 4, drug release in specific parts; 5, odor masking |
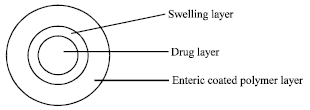 |
| Fig. 6: | Schematic representation of enteric coated system |
Another purpose of enteric coating is drug targeting, as in the case of 5- aminosalicylic acid or the prodrugs mesalazine and sulfasalizine. In these cases, enteric coating is applied such that the drug concentration is increased in the lower parts of the GI tract. Although the use of enteric coating to achieve modified release has been known for a long time, it has always been criticized as to its true value of providing protection and targeted release of the coated active agents.
A survey of the German market showed that more than 50% of enteric formulations were coated with methacrylate copolymers, about 40% with cellulose derivatives, 5%with shellac and 3%with other materials (Kramer et al., 1996). Enteric coating materials (Table 1) are described in various publications (Langguth et al., 1997; Lehmann and Brogmann, 1996). In addition to polymers mentioned in Table 1, others are being studied (e.g., to obtain release at lower pH) (Kokubo et al., 1997). Polymers with dissolution at lower pH are intended for the protection of drugs in acidic medium and not for the protection of the gastric mucosa.
The conclusion of this review is that, from a technical point of view, progress in film-forming polymers, together with advances in excipient technology and modern coating equipment design, has greatly facilitated the design of enteric-coated formulations that fulfill the requirements for controlled and targeted release.
Dosage forms: In general, film-coated dosage forms can be divided into multiple-unit and single unit dosage forms. Single units comprise tablets and film-coated capsules or other forms, usually monolithic structures. Multiple-unit dosage forms can be packages containing granules, capsules containing pellets, or compressed film-coated particles. In the latter situation, total dosage is divided into multiple units that are dispersed in the GI tract, which often results in safer and usually faster action of the drug. Recently, it has also been reported that aqueous dispersions or suspensions can be produced, in which the drug is present in enteric-coated form. The enteric-coated Time Clock System consists of a tablet core coated with a mixture of hydrophobic material and surfactant, which is applied as an aqueous dispersion (Wilding et al., 1994). The drug release from the core of the Time Clock system occurs after a predetermined lag time. This lag time mainly depends on the thickness of the hydrophobic layer and thus is insensitive of GI pH. Investigations that used scintigraphic studies demonstrated that the method for in vitro testing was a good predictor of in vivo release. A greater targeting specificity can be achieved when an enteric coat is additionally applied to this system to avoid problems caused by longer gastric resistance time.
Tablets: Tablets can easily be enteric coated and a variety of products are available on the market, including drugs like acetylsalicylic acid, diclofenac, naproxen (Levien and Baker, 1995), omeprazole, lansoprazole, sodium valproate and many others. Generally, increased bioavailability, improved patient acceptance and formulation stability result from the coating process.
| Table 1: | Properties and applications of enteric coating materials |
 | |
Lehmann investigated the increased stability of acetylsalicylic acid tablets when using enteric film coatings (Lehmann, 1984). Reduction of side effects and increase in patient compliance of enteric-coated acetylsalicylic acid tablets has also been shown in various clinical studies (Orozco-Alcala and Baum, 1979; Bogentoft and Lagerstrom, 1984). In another study (Marvola et al., 1986) different enteric film coatings on pancreatic enzymes were compared. It was found that products containing HPMCP adhered to the gastric mucosa, causing unwanted side effects, including irritation and inflammation of the gastric wall, whereas methacrylic acid copolymers and CAP encountered no such problems. The residence time of the tablets in fed dogs was found to be 6-8 h, which is undesirably long and requires a revised dosage regimen of the tablet (fasted or preprandial).
Capsules: Capsule coating often requires extra precautions (e.g., increased plasticizer content or sometimes an insulating layer), otherwise film coatings or capsule shells may become brittle during storage. Usually the thickness of the film coating layer has to be increased to ensure proper coating of the capsule closure. Vilivalam et al. (2000) demonstrated enteric film coatings with methacrylic copolymers on starch capsules filled with 5-ASA resulted in good storage stability. Good stability was also reported for the enteric coating of hard gelatin capsules containing acetaminophen (Brogmann and Beckert, 2001). Cellulose acetate phthalate was used for an enteric coating on hard gelatin capsules filled with aspirin crystals (Cherrette and Plaizier-Vercammen, 1992). Water uptake into the capsule was found to be unacceptably high, which was attributed to high water vapor permeabilities of cellulose film coatings compared with the more dense methacrylate copolymers. Soft gelatin capsules were also coated with transparent film coatings and good stability on storage was observed (Felton et al., 1995).
MULTIPLE UNITS
A widely used method to produce multiple-unit dosage forms has been the production of sachets that contain film-coated granules. More common is the use of capsules in which enteric-coated particles are filled. A study that used radioactive tracers revealed that enteric-coated erythromycin pellets in capsules were superior to enteric-coated tablets with respect to faster action of the drug caused by a shorter passage time of the coated granules in the stomach (Digenis, 1994; Bechgaard and Ladefoged, 1981; Bechgaard, 1982). In 1998 the first tablet containing enteric-coated particles was marketed (Losec Mups, Omeprazole-Magnesium by ASTRA, Sweden). This is a new principle and may serve as a paradigm of how enteric dosage forms may be designed in the future. However, flexible polymers are required for this purpose and a variety of other factors have to be considered (Beckert et al., 1996; Beckert et al., 1998). In addition to flexibility of the film coating, suitable larger sized filler-binders and stable and strong pellet cores also have to be taken into account. Only the methacrylic acid copolymers seem to have suitable properties necessary to produce these dosage forms. As another example, small microcapsules of ibuprofen were film coated with cellulose acetate phthalate and dispersed in water before administration (Walter et al., 1995). Plasma levels were as expected and did not differ from those of a conventional enteric-coated tablet.
pH-sensitive gels: Many polyanionic materials, such as poly (acrylic acid), are pH sensitive and the degree of swelling of such polymers can be modulated by changing the pH. An application of such technology has been in the development of biomimetic secretory granules for drug delivery applications.
Secretory granules within certain cells consist of a polyanionic polymer network encapsulated within a lipid membrane. The polymer network, which contains biological mediators such as histamine, exists in a collapsed state as a consequence of the internal pH and ionic content which is maintained by the lipid surrounding the granule. Release of histamine from such granules is initiated through the fusion of the granule with the cell membrane exposing the polyanionic internal matrix to the extracellular environment. The change in pH and ionic strength results in ion exchange and swelling of the polyanionic network which in turn causes release of the endogenous mediators.
An environmentally responsive, hydrogel microsphere coated with a lipid bilayer has recently been shown to act as a secretory granule mimic (Fig. 7). Methylene-bis-acrylamide/methacrylic acid anionic microgels were prepared by precipitation polymerization and loaded with doxorubicin and condensed by incubating in buffer at pH 5. The condensed particles were then coated with a lipid bilayer. Disruption of the lipid bilayer by electroporation was shown to cause the microgel particles to swell and release their drug.
The use of these systems in conjunction with temperature-sensitive lipids offers potential to target drugs to areas of inflammation or to achieve site-specific, pulsatile drug delivery through the localized external application of ultrasound or heating to disrupt the lipid bilayers (Hongkee Sah et al., 2001).
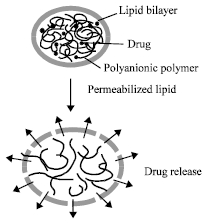 |
| Fig. 7: | A schematic diagram showing the release of drug from a biomimetic secretory granule on disruption of the external lipid bilayer |
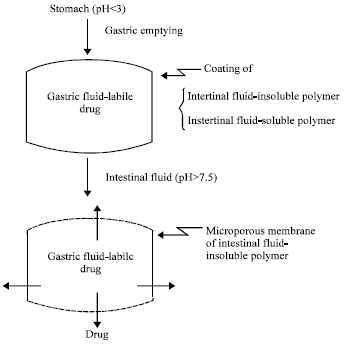 |
| Fig. 8: | Schematic illustration of a pH-activated drug delivery system and the pH-dependent formation of microporous membrane in the intestinal tract |
pH-activated drug delivery systems: For a drug labile to gastric fluid or irritating to gastric mucosa, this type of CrDDS has been developed to target the delivery of the drug only in the intestinal tract, not in the stomach (Chien, 1983). It is fabricated by coating a core tablet of the gastric fluid-sensitive drug with a combination of intestinal fluid-insoluble polymer, like ethyl cellulose and intestinal fluid-soluble polymer, like hydroxylmethyl cellulose phthalate (Fig. 8).
In the stomach, the coating membrane resists the degrading action of gastric fluid (pH<3) and the drug molecules are thus protected from the acidic degradation. After gastric emptying, the CrDDS travels to the small intestine and the intestinal fluid-soluble component in the coating membrane is dissolved away by the intestinal fluid (pH>7.5). This produces a microporous membrane of intestinal fluid-insoluble polymer to control the release of drug from the core tablet. The drug is thus delivered in a controlled manner in the intestine by a combination of drug dissolution in the core and diffusion through the pore channels (Fig. 8). By adjusting the ratio of the intestinal fluid-soluble polymer to the intestinal fluid-insoluble polymer in the membrane, the rate of drug delivery can be regulated. Representative application of this type of CrDDS is in the oral controlled delivery of potassium chloride, which is highly irritating to gastric epithelium. (Chien, 1983).
pH-sensitive liposomes: The concept of pH-sensitive liposomes emerged from the observation that certain enveloped viruses infect cells following acidification of the endosomal lumen to infect cells and from the knowledge that some pathological tissues (tumors, inflamed and infected tissue) have a more acidic environment compared to normal tissues. Although, pH-sensitive liposomes are stable at physiological pH, they destabilize under acidic conditions, leading to the release of their aqueous contents (Ellens et al., 1984; Duzgunes et al., 1983, 1985). In addition, they appear to destabilize or fuse with the membranes of endosomes in which they are internalized, enabling even macromolecular liposome contents to enter the cytoplasm (Straubinger et al., 1983; Straubinger et al., 1985).
The response to acidic pH can be facilitated by a variety of molecules (Duzgunes et al., 1991; Torchilin et al., 1993; Drummond et al., 2000; Venugopalan et al., 2002), including fusogenic peptides incorporated in the lipid bilayer (Parente et al., 1990; Ishiguro et al., 1996; Bailey et al., 1997; Nir et al., 1999; Turk et al., 2002), pH-sensitive lipids (Anderson and Thompson, 1992 Drummond and Daleke, 1995; Reddy and Low, 2000) and pH-sensitive polymers on the surface of liposomes (Leroux et al., 2001; Roux et al., 2002; Mizoue et al., 2002). The combination of phosphatidyl ethanolamine (PE) or its derivatives with molecules with a protonatable group (e.g., carboxylic group) that acts as a stabilizer of PE membranes at neutral pH, is the most commonly used composition. PE has a minimally hydrated and small head group that occupies a lower volume compared to the hydrocarbon chains and can be imagined to have a cone shape, in contrast to the cylinder shape exhibited by phospholipids such a phosphatidylcholine (PC). Strong intermolecular interactions between the amino and phosphate groups of neighboring polar headgroups, along with the cone shape, facilitate the formation of an inverted hexagonal phase at temperatures above a critical temperature (TH) characteristic of the species of PE (Cullis and Kruijff, 1979; Seddon et al., 1983). These properties preclude the preparation of liposomes composed solely of PE or its derivatives under physiological conditions of pH, ionic strength and temperature. Several conditions tend to facilitate the formation of liposomes composed mostly of PE (Simoes et al., 2004): (1) PE can be mixed with other phospholipids, including the zwitterionic PC and the net negatively charged phosphatidylglycerol or phosphatidylserine (PS). These lipids decrease the intermolecular interactions between the polar headgroups of PE and increase the hydration layer of the membrane. (2) High pH (_9.0) confers a net negative charge on PE molecules, due to deprotonation of the amino groups, decreases the intermolecular interactions between the polar headgroups and increases the hydration layer. (3) Amphiphilic molecules containing a protonatable acidic group that is negatively charged at physiological pH can be incorporated alongside PE in the liposome membrane. These molecules not only cause electrostatic repulsion between bilayers, but also disrupt the strong interactions between PE headgroups, thereby allowing the formation of bilayer structures and liposomes at physiological pH and temperature (Peppas and Klier, 1991; Kokubo et al., 1997; Duzgunes et al., 1985; Lai et al., 1985). With this approach, stable liposomes are formed at physiological pH, while at mildly acidic pH the carboxyl groups of the amphiphiles are protonated and their stabilizing effect on PE bilayers is diminished. PE molecules then tend to revert to their inverted hexagonal phase and thus cause liposome destabilization.
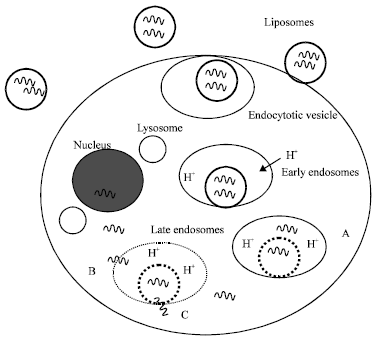 |
| Fig. 9: | Intracellular delivery of oligonucleotides by pH-sensitive liposomes |
Following binding to cells, the liposomes are internalized through the endocytotic pathway. Liposomes are retained in early endosomes that mature into late endosomes. The potential of pH-sensitive liposomes lies in their ability to undergo destabilization at this stage, thus preventing their degradation at the lysosomal level and consequently increasing access to the cytosolic or nuclear targets (Duzgunes et al., 1991; Collins, 1995). Although, non-pH-sensitive liposomes [e.g., containing PC instead of dioleoylphosphatidylethanolamine (DOPE)] are internalized as extensively as pH-sensitive immunoliposomes, their capacity to mediate cytoplasmic delivery of the encapsulated molecules is significantly lower (Straubinger et al., 1985; Ho et al., 1986). This observation suggests that fusion or destabilization of liposomes induced by acidification of the endosomal lumen represents the most important stage in the process of intracellular delivery (Fig. 9).
The liposomes are internalized by endocytosis after binding to cell surface receptors. The lumen of resulting endosomes is acidified by the action of an Hþ-ATPase. The liposomes destabilize at acidic pH, the threshold pH being determined by the composition of the liposomes. The liposomes in the figure have been designed (“programmed”) to destabilize at the lower pH achieved in late endosomes. In case A, the encapsulated oligonucleotides are released into the endosome lumen, but the endosome is not destabilized and thus the contents are trapped in the endosome. In case B, the endosome membrane is also destabilized due to the structural transformation of the pH-sensitive liposomes, enabling the cytoplasmic entry of the oligonucleotides. Alternatively (case C), the liposomes may undergo fusion with the endosome membrane and release their contents directly into the cytoplasm. Some of the oligonucleotides can diffuse into the nucleus.
Studies involving the incubation of cells with lysosomotropic agents (e.g., ammonium chloride or chloroquine) that prevents endosome acidification demonstrate that the efficacy of pH-sensitive liposomes depends on the pH decrease upon endosome maturation. Different molecular mechanisms by which the liposomes release their contents into the cytoplasm have been proposed: (1) destabilization of pH-sensitive liposomes triggers the destabilization of the endosomal membrane, most likely through pore formation, leading to cytoplasmic delivery of their contents; (2) upon liposome destabilization the encapsulated molecules diffuse to the cytoplasm through the endosomal membrane; and (3) fusion between the liposome and the endosomal membranes, leading to cytoplasmic delivery of their contents (Straubinger et al., 1985; Duzgunes et al., 1991; Collins, 1995; Ropert et al., 1993). The fusogenic properties of PE associated with its tendency to form an inverted hexagonal phase under certain conditions favor hypotheses (1) and (3). The fusogenic properties of the liposomes do not always correlate with their efficacy in mediating intracellular delivery. Although aggregation, release of contents and lipid intermixing are observed at low pH with DOPE:cholesteryl hemisuccinate (CHEMS) liposomes, no intermixing of aqueous contents takes place (Ellens et al., 1985), but these liposomes are efficient in delivering their encapsulated contents into cultured cells (Chu et al., 1990). Divalent cations may also play a role in delivery by pH-sensitive liposomes. PE:oleic acid (OA) liposomes undergo fusion in the presence of millimolar concentrations of Ca2þ or Mg2þ and the rate of fusion under acidic conditions is enhanced significantly in the presence of 2 mM Ca2þ (Duzgunes et al., 1985). Cytoplasmic delivery of calcein by DOPE:CHEMS liposomes is inhibited in the presence of ethylenediamine tetraacetic acid (EDTA) (Chu et al., 1990), indicating that divalent cations participate in the destabilization of pH-sensitive liposomes and endosomal membranes, or their fusion with each other.
The efficiency of interaction of pH-sensitive liposomes with cells is sensitive on the inclusion of DOPE in their composition, insensitively of the type of the amphiphilic stabilizer used. In fact, some DOPE-containing liposomes shown to be non-pH-sensitive by biophysical assays, mediated cytoplasmic delivery of their contents as efficiently as well known pH-sensitive formulations (Simoes et al., 2001). Nevertheless, among the different formulations studied, DOPE:CHEMS liposomes had the highest extent of cell association. Results with cells pretreated with metabolic inhibitors or lysosomotropic agents indicate clearly that DOPE-containing liposomes are internalized essentially by endocytosis and that acidification of the endosomes is not the only mechanism involved in the destabilization of the liposomes inside the cell (Simoes et al., 2001). Although some of the liposomes tested had similar abilities to deliver calcein, the delivery of higher molecular weight molecules was highest when encapsulated in pH-sensitive DOPE:CHEMS liposomes compared to other DOPE-containing liposomes (Duzgunes et al., 2001).
Bertrand et al. (2009), characterized the pharmacokinetics (PK) and biodistribution of pH-responsive N-isopropylacrylamide (NIPAAm) copolymers and determined the impact of some physicochemical parameters on their biological profiles. Radiolabeled copolymers of NIPAAm and methacrylic acid (MAA) of different molecular weight, amphiphilicity and lower critical solution temperature (LCST) were synthesized and injected intravenously to rats. The PK and excretion profiles were monitored over 48 h. It was found that elimination occurred mainly through urinary excretion, which was principally governed by molecular weight The polymers with an LCST situated below the physiological temperature did not circulate for prolonged periods in the bloodstream and were highly captured by the organs of the mononuclear phagocyte system. Finally, the complexation of an alkylated pH-sensitive polymer with a molecular weight of 10, 000 to the bilayer of PEGylated liposomes produced a drastic change in the PK parameters, indicating that the polymer remained anchored in the phospholipid bilayer in the bloodstream. These data indicate that stable pH-sensitive liposomes can be produced using excretable NIPAAm copolymers.
Yuba et al. (2010) demonstrated that these linear polymer-modified liposomes exhibited a pH-dependent membrane fusion behavior in cellular acidic compartments. They investigated the backbone structure to obtain pH-sensitive polymers with much higher fusogenic activity and to reveal the effect of the polymer backbone structure on the interaction with the membrane. Hyperbranched poly(glycidol) (HPG) derivatives were prepared as a new type of pH-sensitive polymer and used for the modification of liposomes. HPG derivatives showed a stronger interaction with the membrane than the linear polymers show. Liposomes modified with HPG derivatives of high DP delivered contents into the cytosol of DC 2.4 cells, a dendritic cell line, more effectively than the linear polymer-modified liposomes do. Results show that the backbone structure of pH-sensitive polymers affected their pH-sensitivity and interaction with liposomal and cellular membranes.
pH sensitive microspheres: The pH of the human gastrointestinal tract was shown to increase progressively from the stomach (pH 2-3), small intestine (pH 6.5- 7) to the colon (7.0-7.8) (Ashford and Fell, 1994). Recent studies using sensitive and reliable equipment have given more exact data showing that the pH values in the stomach range from 1.2 to 5.0, while the pH values in the duodenum, jejunum and ileum and colon are 6.6±0.5, 7.4±0.4, 7.5±0.4 and 7.0±0.7, respectively (Fallingborg, 1999; Russell et al., 1993; Song et al., 2002). It has also been reported in many articles that the average gastric emptying time of multiple units was in the range 1-3 h in a fasted state and 2-4 h in a fed state. The small intestinal transit is surprisingly constant at 3-4 h and appears to be insensitive of the type of dosage form and whether the subject is in the fasted or fed state (Davis et al., 1986; Watts and Illum, 1997). Therefore, a dosage form could take from as little as 4 h to longer than 8 h to arrive at the colon following oral administration. It was found that the changes in the pH of the gastrointestinal tract had a certain gradient and the transit time of materials through the gut was comparatively long. Moreover, many pH-sensitive polymers (Table 2) such as Eudragit E, Eudragit L, Eudragit S, HP-55 and CAP, etc., which could dissolve at different pH values, have been synthesized and exploited widely in designing dosage forms. These findings provided the foundation for designing our pH-sensitive drug delivery system. Since the drug release persists throughout the whole gastrointestinal tract, this results in sustained transport of the drug and a prolonging of its pharmacological action in vivo. Only part of the formulated drug was released from the system at different locations in the gastrointestinal tract, the peak and valley phenomenon of conventional formulations could be avoided and the side effects of the drug could also be reduced.
Based on the above consideration Yang et al. (2004), developed pH-dependent delivery system of nitrendipine in which they have mixed three kinds of pH dependent microspheres made up of acrylic resins Eudragit E-100, Hydroxypropylmethylcellulose phthalate and Hydroxypropylmethylcellulose acetate succinate as pH sensitive polymers. In one of the study carried out by Mastiholimath et al. (2007) attempt was made to deliver theophylline into colon by taking the advantage of the fact that colon has a lower pH value (6.8) than that of the small intestine (7.0-7.8). So, by using the mixture of the polymers, i.e., Eudragit L and Eudragit S in proper proportion, pH sensitive release in the colon was obtained.
| Table 2: | pH sensitive polymer with their threshold pH |
 | |
pH sensitive nanoparticles: Indeed, it was recently reported that particles in the size range 40-120 nm were translocated both transcellularly and paracellularly (Mathiowitz et al., 1997). In addition to the potential for enhancing drug bioavailability via particle uptake mechanisms, particulate oral delivery systems can protect labile macromolecules from stomach acid and from the first-pass metabolism in the gastrointestinal tract. Likewise, particulate formulations also can increase transit times than larger dosage forms and can increase the local concentration gradient across absorptive cells. Thereby enhancing local and systemic delivery or both free and bound drugs across the gut (Kreuter et al., 1989). Previous studies have described the use of pH sensitive polymers such as hydroxypropylmethylcellulose phthalate (Klipstein et al., 1983), Eudragit® L100 and Eudragit® S100 (Morishita et al., 1993; Jaeghere et al., 2000) or cellulose acetate phthalate (Lin et al., 1991) to encapsulate antigens or proteins for oral administration. These pH-sensitive particles are matrix-type dispersed systems. Release of the highly dispersed drug at a specific pH within the gastrointestinal tract, as close as possible to the absorption window of the drug, is expected to increase the probability of drug absorption and to minimize the first-pass metabolism of drug.
On the basis of the above mentioned considerations, Dai et al. (2004) were thought plausible to combine the advantages of nanoparticles as oral delivery systems with the benefits of the pH-sensitive property.
Lu et al. (2008) prepared pH-sensitive nanoparticle drug delivery system derived from natural polysaccharide pullulan for doxorubicin (DOX) release. Pullulan was functionalized by successive carboxymethylization and amidation to introduce hydrazide groups. DOX was then grafted onto pullulan backbone through the pH-sensitive hydrazone bond to form a pullulan/DOX conjugate. This conjugate self-assembled to form nano-sized particles in aqueous solution as a result of the hydrophobic interaction of the DOX. Transmission Electron Microscope (TEM) and Dynamic Light Scattering (DLS) characterization showed that the nanoparticles were spherical and their size was less than 100 nm. The DOX released from the nanoparticles in a pH-sensitive manner.
Methods of preparing of polymeric nanoparticles (Soppimath et al., 2001; Mohanraj and Chen, 2006) include ionic gelation, coacervation, solvent evaporation, spontaneous emulsification/solvent diffusion, salting out/emulsification-diffusion, supercritical fluid technology and polymerization.
CONCLUSION
Presently, oral delivery of drug is still by far the most preferable route of drug delivery due to the ease of administration, patient compliance and flexibility in its formulations. Generally, sustained and controlled-release products provide a desired therapeutic effect, but fall short of diseases following biological rhythms. Circadian disorders such as hypertension, osteoarthritis, asthma etc., which require chronopharmacotherapy. PSDDS can effectively tackle this problem as it is modulated according to body's circadian clock giving release of drug. Multiparticulate systems are useful for treatment of patients; due to their resulting high efficiency and robustness. There are various technologies present in the market based on the various methodolgies. pH sensitive release systems should be promising in the future.
ACKNOWLEDGMENT
The author Mr. Balamuralidhara V. is highly thankful to Dr. H.G. Shivakumar, Principal, JSS College of Pharmacy, for his constant encouragement and support and JSS University, Mysore, for providing the required infrastructure to carryout the project on the pH sensitive drug delivery systems. We thank Ms Srirupa Biswas for helping in the preparation of the manuscript.
REFERENCES
- Yoshida, R., K. Sakai, T. Okano and Y. Sakurai, 1993. Pulsatile drug delivery systems using hydrogels. Adv. Drug Delivery Rev., 11: 85-108.
CrossRefDirect Link - Brannon-Peppas, L. and N.A. Peppas, 1989. Solute and penetrant diffusion in swellable polymers. IX. The mechanisms of drug release from ph-sensitive swelling-controlled systems. J. Controlled Release, 8: 267-274.
CrossRefDirect Link - Annaka, M. and T. Tanaka, 1992. Multiple phases of polymer gels. Nature, 355: 430-432.
CrossRefDirect Link - Firestone B.A. and R.A. Siegel, 1988. Dynamic pH-dependent swelling properties of a hydrophobic polyelectrolyte gel. Polym. Commun., 29: 204-208.
Direct Link - Kou, J.H., D. Fleisher and G.L. Amidon, 1990. Modeling drug release from dynamically swelling poly(hydroxymethyl methacrylate-co-methacrylic acid) hydrogels. J. Controlled Release, 12: 241-250.
CrossRefDirect Link - Pradny, M. and J. Kopecek, 1990. Hydrogels for site-specific oral delivery. Poly[(acrylic acid)-co-(butyl acrylate)] crosslinked with 4,4-di(methacryl amino) azobenzene. Makromol. Chem., 191: 1887-1897.
CrossRefDirect Link - Kono, K., F. Tabata and T. Takagishi, 1993. pH-responsive permeability of poly(acrylic acid)-poly(ethylenimine) complex capsule membrane. J. Membr. Sci., 76: 233-243.
CrossRefDirect Link - Hariharan, D. and N.A. Peppas, 1993. Modelling of water transport and solute release in physiologically sensitive gels. J. Contr. Rel., 23: 123-135.
CrossRefDirect Link - Siegel, R.A. and B.A. Firestone, 1988. pH-dependent equilibrium swelling properties of hydrophobic polyelectrolyte copolymer gels. Macromolecules, 21: 3254-3261.
CrossRefDirect Link - Allcock, H.R. and A.M. Ambrosio, 1996. Synthesis and characterization of pH-sensitive poly(organophosphazene) hydrogels. Biomaterials, 17: 2295-2302.
CrossRefPubMedDirect Link - Bell, C. and N. Peppas, 1996. Water, solute and protein diffusion in physiologically responsive hydrogels of poly(methacrylic acid-g-ethylene glycol). Biomaterials, 17: 1201-1218.
CrossRefPubMedDirect Link - Jarvinen, K., S. Akerman, B. Svarfvar, T. Tarvainen, P. Vlinikka and P. Paronen, 1998. Drug release from pH and ionic strength responsive poly(acrylic acid) grafted poly(vinylidenefluoride) membrane bags in vitro. Pharm. Res., 15: 802-805.
CrossRefDirect Link - Siegel, R.A., I. Johannes, A. Hunt and B.A. Firestone, 1992. Buffer effects on swelling kinetics in polybasic gels. Pharm. Res., 9: 76-81.
CrossRefPubMedDirect Link - Giannos, S., S. Dinh and B. Berner, 1995. Temporally controlled drug delivery systems: Coupling of pH oscillators with membrane diffusion. J. Pharm. Sci., 84: 539-543.
CrossRefPubMedDirect Link - Heller, J. and P.V. Trescony, 1979. Controlled drug release by polymer dissolution II: Enzyme-mediated delivery device. J. Pharm. Sci., 68: 919-921.
CrossRefPubMedDirect Link - Ghandehari, H., P. Kopeckova and J. Kopecek, 1997. In vitro degradation of pH-sensitive hydrogels containing aromatic azo bonds. Biomaterials, 18: 861-872.
CrossRefPubMedDirect Link - Petka, W.A., J.L. Harden, K.P. McGrath, D. Wirtz and D.A. Tirrell, 1998. Reversible hydrogels from self-assembling artificial proteins. Science, 281: 389-392.
CrossRefPubMedDirect Link - Li, J.J., 2003. Circadian variation in myocardial ischemia: The possible mechanisms involving in this phenomenon. Med. Hypotheses, 61: 240-243.
CrossRefPubMedDirect Link - Lamberg, L., 1991. Chronotherapeutics: Implications for drug therapy: When patients take medications may be as important as what they take. Am. Pharmacy, 31: 20-23.
CrossRefPubMedDirect Link - Firestone, B.A. and R.A. Siegel, 1991. Kinetics and mechanisms of water sorption in hydrophobic, ionizable copolymer gels. J. Appl. Polym. Sci, 43: 901-914.
CrossRefDirect Link - Falamarzian, M. and J. Varshosaz, 1998. The effect of structural changes on swelling kinetics of polybasic/hydrophobic pH-sensitive hydrogels. Drug Dev. Ind. Pharm., 24: 667-669.
CrossRefPubMedDirect Link - Kou, J.H., G.L. Amidon and P.I. Lee, 1988. pH-dependent swelling and solute diffusion characteristics of poly(hydroxyethyl methacrylate-co-methacrylic acid) hydrogels. Pharm. Res., 5: 592-597.
CrossRefPubMedDirect Link - Brannon-Peppas, L. And N.A. Peppas, 1990. Dynamic and equilibrium swelling behaviour of pH-sensitive hydrogels containing 2-transhydroxyethyl methacrylate. Biomaterials, 11: 635-644.
CrossRefPubMedDirect Link - Khare, A.R. and N.A. Peppas, 1993. Release behavior of bioactive agents from pH-sensitive hydrogels. J. Biomater. Sci. Poly, 4: 275-289.
CrossRefDirect Link - Peppas, N.A. and J. Klier, 1991. Controlled release by using poly(methacrylic acid-g-ethylene glycol) hydrogels. J. Controlled Release, 16: 203-214.
CrossRefDirect Link - Siegel, R.A., M. Falamarzian, B.A. Firestone and B.C. Moxley, 1988. pH-controlled release from hydrophobic/polyelectrolyte copolymer hydrogels. J. Controlled Release, 8: 179-182.
CrossRefDirect Link - Patel, V.R. and M.M. Amiji, 1996. Preparation and characterization of freeze-dried chitosan-poly(ethylene oxide) hydrogels for site-specific antibiotic delivery in the stomach. Pharm. Res., 13: 588-593.
CrossRefPubMedDirect Link - Akala, E.O., P. Kopeckova and J. Kopecek, 1998. Novel pH-sensitive hydrogels with adjustable swelling kinetics. Biomaterials, 19: 1037-1047.
CrossRefPubMedDirect Link - Gutowska, A., J.S. Bark, I.C. Kwon, Y.H. Bae and S.W. Kim, 1997. Squeezing hydrogels for controlled oral drug delivery. J. Controlled Release, 48: 141-148.
CrossRefDirect Link - Bilia, A., V. Carelli, G. Di Colo and E. Nannipieri, 1996. In vitro evaluation of a pH-sensitive hydrogel for control of GI drug delivery from silicone-based matrices. Int. J. Pharm., 130: 83-92.
CrossRefDirect Link - Carelli, V., S. Coltelli, G. Di Colo, E. Nannipieri and M.F. Serafini, 1999. Silicone microspheres for pH-controlled gastrointestinal drug delivery. Int. J. Pharm., 179: 73-83.
CrossRefPubMedDirect Link - Aikawa, K., K. Matsumoto, H. Uda, S. Tanaka and S. Tsuchiya, 1998. Hydrogel formation of the pH response polymer polyvinylacetal diethylaminoacetate (AEA). Int. J. Pharm., 167: 97-104.
CrossRefDirect Link - Aikawa, K., K. Matsumoto, H. Uda, S. Tanaka and S. Tsuchiya, 1998. Drug release from pH-response polyvinylacetal diethylaminoacetate hydrogel, and application to nasal delivery. Int. J. Pharm., 168: 181-188.
CrossRefDirect Link - Dong, L.C. and A.S. Hoffman, 1990. Synthesis and application of thermally reversible heterogels for drug delivery. J. Controlled Release, 13: 21-31.
CrossRefDirect Link - Dong, L.C. and A.S. Hoffman, 1991. A novel approach for preparation of pH-sensitive hydrogels for enteric drug delivery. J. Controlled Release, 15: 141-152.
CrossRefDirect Link - Vakkalanka, S.K., C.S. Brazel and N.A. Peppas, 1997. Temperature- and pH-sensitive terpolymers for modulated delivery of streptokinase. J. Biomater. Sci. Poly, 8: 119-129.
CrossRefPubMedDirect Link - Brazel, C.S. and N.A. Peppas, 1996. Pulsatile local delivery of thrombolytic and antithrombotic agents using poly(N-isopropylacrylamide-co-methacrylic acid) hydrogels. J. Controlled Release, 39: 57-64.
CrossRefDirect Link - Chiu, H.C., G.H. Hsiue, Y.P. Lee and L.W. Huang, 1999. Synthesis and characterization of pH-sensitive dextran hydrogels as a potential colon-specific drug delivery system. J. Biomater. Sci. Poly, 10: 591-608.
CrossRefPubMedDirect Link - Markland, P., Y. Zhang, G.L. Amidon and V.C. Yang, 1999. A pH- and ionic strength-responsive polypeptide hydrogel: Synthesis, characterization, and preliminary protein release studies. J. Biomed. Mater. Res., 47: 595-602.
CrossRefPubMedDirect Link - Langguth, P., V. Bohner, J. Heizmann, H.P. Merkle, S. Wolffram, G.L. Amidon and S. Yamashita, 1997. The challenge of proteolytic enzymes in intestinal peptide delivery. J. Controlled Release, 46: 39-57.
CrossRefDirect Link - Kokubo, H., S. Obara, K. Minemura and T. Tanaka, 1997. Development of cellulose derivatives as novel enteric coating agents soluble at pH 3.5-4.5 and higher. Chem. Pharm. Bull., 45: 1350-1353.
CrossRefPubMedDirect Link - Wilding, R., S.S. Davis, F. Pozzi, P. Furlani and A. Gazzaniga, 1994. Enteric coated timed release systems for colonic targeting. Int. J. Pharmaceutics, 111: 99-102.
CrossRefDirect Link - Levien, T. and D. Baker, 1995. Reviews of naproxen enteric coated and varicella vaccine. Hospital Pharm., 30: 1011-1024.
CrossRef - Bogentoft, E.B. and P.O. Lagerstrom, 1984. Comparison of two enteric-coated acetylsalicylic acid preparations by monitoring steady state levels of salicylic acid and its metabolites in plasma and urine. Biopharm. Drug Dispos., 5: 251-260.
PubMed - Vilivalam,V., L.L. Illum and K.K. Iqbal, 2000. Starch capsules: An alternative system for oral drug delivery. Pharm. Sci. Technol. Today, 3: 64-69.
PubMed - Felton, L.A., M.M. Haase, N.H. Shah, G. Zhang and M.H. Infeld et al., 1995. Physical and enteric properties of soft gelatin capsules coated with Eudragit L30 D-55. Int. J. Pharm., 113: 17-24.
CrossRef - Bechgaard, H. and K. Ladefoged, 1981. Gastrointestinal transit time of single-unit tablets. J. Pharm. Pharmacol., 33: 791-792.
PubMed - Beckert, T., K. Lehmann and P.C. Schmidt, 1996. Compression of enteric-coated pellets to disintegrating tablets. Int. J. Pharm., 143: 13-23.
CrossRef - Walter, K., G. Weib, A. Laicher and F. Stanislaus, 1995. Pharmacokinetics of ibuprofen following single administration of a suspension containing enteric-coated microcapsules. Drug Res., 45: 886-890.
PubMed - Ellens, H., J. Bentz and F.C. Szoka, 1984. pH-induced destabilization of phosphatidyl ethanolamine-containing liposomes: Role of bilayer contact. Biochemistry, 23: 1532-1538.
CrossRef - Torchilin, V.P., F. Zhou and L. Huang, 1993. pH-Sensitive liposomes. J. Liposome Res., 3: 201-255.
CrossRef - Drummond, D.C., M. Zignani and J. Leroux, 2000. Current status of pH-sensitive liposomes in drug delivery. Prog. Lipid Res., 39: 409-460.
PubMed - Venugopalan, P., S. Jain, S. Sankar, P. Singh, A. Rawat and S.P. Vayas, 2002. pH-sensitive liposomes: Mechanism of triggered release to drug and gene delivery prospects. Pharmazie, 57: 659-671.
PubMed - Ishiguro, R., M. Matsumoto and S. Takahashi, 1996. Interaction of fusogenic synthetic peptide with phospholipid bilayers: Orientation of the peptide alpha-helix and binding isotherm. Biochemistry, 35: 4976-4983.
PubMed - Leroux, J., E. Roux, D. Le Garrec, K. Hong and D.C. Drummond, 2001. N-isopropylacrylamide copolymers for the preparation of pH-sensitive liposomes and polymeric micelles. J. Control Releases, 72: 71-84.
PubMed - Cullis, P.R. and B. De Kruijff, 1979. Lipid polymorphism and the function role of lipids in biological membranes. Biochim. Biophys. Acta, 559: 399-420.
PubMed - Seddon, J.M., G. Cevc and D. Marsh, 1983. Calorimetric studies of the gel-fluid (Lβ-La) and lamellar-inverted hexagonal phase (La-HII) transition in dialkyl-and diacylphosphatidyl ethanolamine. Biochemistry, 22: 1280-1289.
CrossRef - Simoes, S., J.N. Moreira, C. Fonseca, N. Duzgunes and M.C. de Lima, 2004. On the formulation of pH-sensitive liposomes with long circulation times. Adv. Drug Deliv. Rev., 56: 947-965.
PubMed - Ellens, H., J. Bentz and F.C. Szoka, 1985. H+- and Ca2+-induced fusion and destabilization of liposomes. Biochemistry, 24: 3099-3106.
PubMed - Duzgunes, N., S. Simoes, V. Slepushkin, E. Pretzerb and J.J. Rossi et al., 2001. Enhanced inhibition of HIV-1 replication in macrophages by antisense oligonucleotides, ribozymes and acyclic nucleoside phosphonate analogs delivered in pH-sensitive liposomes. Nucleos Nucleot Nucl Acids, 20: 515-523.
CrossRefPubMed - Fallingborg, J., 1999. Intraluminal pH of the human gastrointestinal tract. Dan. Med. Bull., 46: 183-196.
PubMed - Mathiowitz, E., J.S. Jacob, Y.S. Jong, G.P. Carino and D.E. Chickering et al., 1997. Biologically erodable microspheres as potential oral delivery systems. Nature, 386: 410-414.
PubMed - Kreuter, J., U. Muller and K. Munz, 1989. Quantitative and microautoradiographic study on mouse intestinal distribution of polycyanoacrylate nanoparticles. Int. J. Pharm., 55: 39-45.
CrossRef - Klipstein, F.A., R.F. Engert and W.T. Sherman, 1983. Peroral immunization with Escherichia coli heat-labile enterotoxin delivered by microspheres. Infect. Immun., 39: 1000-1003.
PubMed - Morishita, I., M. Morishita, K. Takayama, Y. Machida and T. Nagai, 1993. Enteral insulin delivery by microspheres in 3 different formulations using Eudragit L100 and S100. Int. J. Pharm., 91: 29-37.
CrossRefDirect Link - Jaeghere, F.D., E. Allemann, F. Kubel, B. Galli, R. Cozens and E. Doelker, 2000. Oral bioavailability of a poorly water soluble HIV-1 protease inhibitor incorporated into pH-sensitive particles: Effect of the particle size and nutritional state. J. Controlled Release, 68: 291-298.
CrossRef - Lin, S.Y., Y.L. Tzan, C.J. Lee and C.N. Weng, 1991. Preparation of enteric coated microspheres of Mycoplasma hyopneumoniae vaccine with cellulose acetate phthalate. I. Formation condition and micromeritic properties. J. Microencapsule, 8: 317-325.
CrossRef - Dai, J., T. Nagai, X. Wang, T. Zhang, M. Menga and Q. Zhang, 2004. pH-sensitive nanoparticles for improving the oral bioavailability of cyclosporine A. Int. J. Pharmaceut., 280: 229-240.
CrossRefPubMedDirect Link - Soppimath, K.S., T.M. Aminabhavi, A.R. Kulkarni and W.E. Rudzinski, 2001. Biodegradable polymeric nanoparticles as drug delivery devices. J. Controlled Release, 70: 1-20.
CrossRefPubMedDirect Link - Mastiholimath, V.S., P.M. Dandagi, S.S. Jain, A.P. Gadad and A.R. Kulkarni, 2007. Time and pH dependent colon specific, pulsatile delivery of theophylline for nocturnal asthma. Int. J. Pharm., 328: 49-56.
PubMed - Yang, M., F. Cui, B. You, J. You, L. Wang, L. Zhang and Y. Kawashima, 2004. A novel pH-dependent gradient-release delivery system for nitrendipine II Investigations of the factors affecting the release behaviors of the system. Int. J. Pharm., 286: 99-109.
PubMed - Lu, D.X., X.T. Wen, J. Liang, X.D. Zhang, Z.W. Gu and Y.J. Fan, 2008. Novel pH-sensitive drug delivery system based on natural polysaccharide for doxorubicin release. Chinese J. Polymer Sci., 26: 369-374.
CrossRef - Yuba, E., A. Harada, Y. Sakanishi and K. Kono, 2010. Carboxylated hyperbranched poly(glycidol)s for preparation of pH-sensitive liposomes. J. Controlled Release.
CrossRef - Yang, L., J.S. Chu and J.A. Fix, 2002. Colon-specific drug delivery: New approaches and in vitro/in vivo evaluation. Int. J. Pharma., 235: 10-15.
CrossRef - Vishal Gupta, N., C.S. Satish and H.G. Shivakumar, 2007. Preparation and characterization of gelatin-poly(methacrylic acid) IPN hydrogels as a pH-sensitive delivery system for glipizide. Indian J. Pharm. Sci., 69: 64-68.
CrossRef - Vishal Gupta, N. and H.G. Shivakumar, 2009. Preparation and characterization of superporous hydrogels as a pH-sensitive drug delivery system for pantoprazole sodium. Curr. Drug Deliv., 6: 505-510.
PubMed - Mohanraj, V.J. and Y. Chen, 2006. Nanoparticles: A review. Trop. J. Pharma. Res., 5: 561-573.
Direct Link - Beckert, T., K. Lehmann and P.C. Schmidt, 1998. Compression of enteric-coated pellets to disintegrating tablets: Uniformity of dosage units. Powder Tech., 96: 248-254.
CrossRef