
Jayesh Anerao
National Facility for Biopharmaceuticals, G.N. Khalsa College, Matunga, 19 Mumbai, India
LiveDNA: 91.8005
Vikas Jha
National Facility for Biopharmaceuticals, G.N. Khalsa College, Matunga, 19 Mumbai, India
Nitin Desai
Amity Institute of Biotechnology, Amity University Mumbai, 410206 Panvel, India
Asian Journal of Biotechnology
Year: 2017 | Volume: 9 | Issue: 1 | Page No.: 35-42







ABSTRACT
Background: Genetic analysis and DNA barcoding technology of plant relies on high yields of pure DNA samples. The DNA barcoding has the potential to provide an alternative means of estimating species richness without high level expertise in field identification skills and in a much shorter time frame. Western Ghats of India is one of the mega diversity centres in the world. There are 15 species of Garcinia reported of which many are endemic in nature. Genus Garcinia store large amounts of phenolics, polysaccharides, polyphenols, tannins and other metabolites within their leaf tissue making genomic DNA extraction difficult. While many DNA extraction methods exist that contend with the presence of phenolics and polysaccharides, these methods rely mainly on selection of material. Materials and Methods: The DNA of G. indica and G. xanthochymus was extracted by using DNAzol® kit, modified SDS and C-TAB methods. A modified C-TAB and SDS method was optimized for removal of polyphenolics. Results: The protocol developed yielded 2000-2500 ng μL–1 of quality DNA without any impurities as evident by A260/280 ratio ranging from 1.78-1.81 and 260/230 ratio ranging from 2.15-2.18 for 3 g of the leaf tissue. The extraction of DNA by SDS was most effective from young leaves. In this study the properties of Triton X in the second purification step to remove lipid and protein component of the cellular membranes has been exploited. Triton X helps in decreasing the surface tension and along with that it helps in solubilization of nonpolar entities. High Concentration of LiCl2 (0.5 M) in presence of ethanol helps in salting out of DNA. The resulted genomic DNA showed fine Random Amplified Polymorphic DNA (RAPD) Inter Simple Sequence Repeat (ISSR) banding pattern. The DNA obtained was also amenable to rbcL barcode gene amplification of plant. Conclusion: The protocol optimized is reproducible and useful for other members of Garcinia species.
PDF Abstract XML References Citation
Received: July 20, 2016;
Accepted: November 14, 2016;
Published: December 15, 2016
How to cite this article
Jayesh Anerao, Vikas Jha and Nitin Desai, 2017. Optimization of DNA Extraction Methods from Garcinia species for ISSR-PCR, RAPD-PCR and DNA Barcoding. Asian Journal of Biotechnology, 9: 35-42.
DOI: 10.3923/ajbkr.2017.35.42
URL: https://scialert.net/abstract/?doi=ajbkr.2017.35.42
DOI: 10.3923/ajbkr.2017.35.42
URL: https://scialert.net/abstract/?doi=ajbkr.2017.35.42
INTRODUCTION
The genus Garcinia L., belongs to the family Clusiaceae (Guttiferae). This family consists of approximately 200 species throughout the world, among which 36 species occur in India. The Western Ghats region is considered as a secondary center of origin for Garcinia species, where 6 species are endemic to Western Ghats1. Prominent characteristics of genus Garcinia are monopodial growth, coriaceous texture of leaves, oil glands or cavities containing yellow or bright coloured resins present in all parts of the plants and polygamodioecious nature. Garcinia mangostana is known as the queen of fruits and is well studied for morphological variability, flowering method, fertilization and fruit variation2. Garcinia atroviridis is a well-studied endemic species of Malaysia3.
Genetic studies give direct knowledge of the gene variations at intergeneric as well as intrageneric level. Molecular markers provide an opportunity for direct comparison and identification of different genetic material. To see this variation, DNA isolation protocols need to be optimized. Extraction and isolation of pure and high quality DNA is essential for any molecular studies, however, DNA extraction from any tree species is usually compromised becuase of excess amounts of polyphenols, tannins and other secondary metabolites. So, the DNA isolation methods have to be optimized for each plant species and their components because of the presence of these secondary metabolites4. These compounds always interfere with DNA isolation and its purity5. To date, various DNA isolation protocols have been reported including kit based methods, still DNA isolation is a challenge for many plant species.
An ideal extraction technique should optimize the DNA yield, minimize DNA degradation and be efficient in terms of cost, time, labor and supplies. It must also be suitable for extracting multiple samples and generate minimal hazardous waste.
Present study deals with solving this issue by isolating the total genomic DNA from young leaves of Indian Garcinia species. The method described herein given an optimized protocol for isolation of high quality of genomic DNA, free of contamination. This study also provides RAPD-PCR, ISSR-PCR and barcoding studied in G. indica, G. xanthochymus.
MATERIALS AND METHODS
Plant material: Fruits and seeds of G. indica and G. xanthochymus were collected from Dapoli and Devrukh region (Maharashtra state). The seeds obtained were germinated and plants were mainteind at botanical garden of G.N. Khalsa college (Mumbai). The GPS locations of collected samples is given in Table 1. Young leaves of Garcinia plants were collected prior to extraction of DNA.
Extraction of DNA
DNA extraction by plant DNAzol® kit: DNAzol® involves a single extraction buffer that solubilizes all cellular components and allows selective precipitation of DNA in the presence of ethanol. The first protocol used a commercially available plant DNAzol® kit and the manufacturer’s instructions were followed. Briefly 3 g of G. indica, G. xanthochymus and onion peels were taken as control and ground in 9 mL of DNAzol® kit buffer. The samples were incubated at 60°C for 1 h. Equal volume of chloroform was added and mixed with it properly. The samples were centrifuged at 10,000 rpm for 10 min. The supernatant was separated and the step was repeated. Chloroform washing helps in removal of contaminants. The upper aqueous layer was then separated and chilled ethanol was added to precipitate the DNA. The samples were kept in -20°C for 45 min. After precipitation of DNA, the samples were centrifuged and the supernatant was discarded. The pellet was given a wash with 70% ethanol and centrifuged at 10,000 rpm for 10 min. Finally absolute ethanol was added and centrifuged at 10,000 rpm for 10 min. The pellet was kept for drying at 42°C and then resuspended in 50 μL of TE buffer.
DNA extraction by C-TAB (Cetyl-trimethyl ammonium bromide) protocol: The DNA extraction procedure was performed according Doyle and Doyle6 method with slight modifications. The G. indica, G. xanthochymus and onion peels weighing 3 g were homogenized in 9 mL of C-TAB buffer and incubated at 60°C for 1 h. After 1 h the samples were centrifuged at 10,000 rpm for 10 min. The supernatant was treated with equal volume of chloroform:isoamyl alcohol (24:1) and centrifuged at 10,000 rpm for 10 min. The above step was repeated to increase the level of purity.
| Table 1: | Details of collection sites and its geographical locations of Garcinia species |
 | |
The aqueous layer was aliquoted and used for precipitation of DNA by adding two volumes of chilled ethanol. The samples were incubated at -20°C for 45 min and centrifuged at 10,000 rpm for 10 min. The pellet was washed with 70% ethanol and centrifuged at 10,000 rpm for 10 min. The pellet obtained was washed with absolute ethanol and centrifuged at 10,000 rpm for 10 min. The pellet was kept for drying at 42°C and resuspended in 50 μL of TE buffer.
DNA extraction by SDS (Sodium dodecyl sulphate) protocol: The SDS method reported by Dellaporta et al.7 with slight modifications was performed on G. indica, G. xanthochymus and onion peels. About 3 g of fresh leaflets were ground in 9 mL of SDS extraction buffer. The mixture was incubated at 60°C for 1 h. Then the samples were centrifuged at 10,000 rpm for 10 min and the supernatant was treated with equal volumes of chloroform:isoamyl alcohol twice to get a clear interface. The mixture was centrifuge at 10,000 rpm for 10 min. The supernatant was transferred into a new tube and double volumes of chilled absolute ethanol was added to it. The samples were kept for incubation at -20°C for 45 min to allow the DNA to precipitate and later centrifuged at 10,000 rpm at 4°C for 10 min. The DNA pellet was given a wash with 70% ethanol and centrifuged at 10,000 rpm for 10 min. The wash with absolute ethanol was repeated and the pellet was kept for drying at 42°C. Once dried, the pellet was resuspended in 50 μL TE buffer.
Purification of DNA: The resuspended DNA samples were pooled in a single eppendorf tube for each species. The 0.1 μg mL–1 RNAse A was added in each sample tube to remove RNA contamination. The tubes were kept for incubation at 55°C for 30 min 1X Triton X was added from 10X stock solution. The solution was mixed gently and treated with equal volume of chloroform. The solution was centrifuged at 4°C for 10 min at 10,000 rpm. The aqueous phase was aliquoted and 1/10th of 5 M LiCl2 along with 1 mL of chilled ethanol was added into it. The DNA appeared in white spool. The solution was kept for incubation at -20°C for 45 min and later centrifuged at 10,000 rpm for 10 min. The pellet was given a wash with 70% ethanol followed by a wash with absolute ethanol and kept for air drying. After drying, the pellet was resuspended in 50 μL TE buffer.
Quantification and visualization of DNA: The concentration and purity of DNA was checked on Nanodrop spectrophotometer. The absorbance at 260 nm showed sharp peaks of the extracted DNA.
| Table 2: | RAPD, ISSR and rbcL primers used for amplification of DNA |
 | |
The DNA was visualized by running on 0.8% Agarose gel stained with 0.1 μg mL–1 ethidium bromide.
PCR amplification: The PCR was carried out in 25 μL volume reaction mixture. The reaction mixture contained 100 ng of DNA template, 2.5 U Taq polymerase enzyme (Thermofischer), 0.2 mM dNTPs, 2.5 mM MgCl2, 1X Taq DNA polymerase buffer and 0.5 μM primers (Euroffins, India). For Inter Simple Sequence Repeats (ISSR) and Random Amplification of Polymorphic DNA (RAPD) primers (Table 2), the DNA was amplified using the following conditions with initial denaturation at 95°C for 5 min, followed by 45 cycles of denaturation at 94°C for 1 min, annealing temperature at 32°C for RAPD and 52°C for ISSR for 1 min and extension at 72°C for 1 min followed by final extension at 72°C for 7 min. For DNA barcoding primer rbcL (ribulose-1,5-bisphosphate carboxylase/oxygenase large subunit, 613 bp amplicon length) the PCR set up was as follows. Initial denaturation at 95°C for 5 min, followed by 35 cycles of denaturation at 94°C for 1 min, annealing temperature at 58°C for 1 min and extension at 72°C for 1 min followed by final extension at 72°C for 10 min. The PCR products were separated on 2% agarose gel stained with 0.1 μg mL–1 ethidium bromide, using 1X TAE buffer.
RESULTS
Selection of the material: Selection of the leaf tissue is very important for DNA extraction from woody tree species. In this study, the leaf tissue was harvested from very young seedlings and used for DNA extraction because fresh, tender leaf tissue was preferable as they contain less polyphenols and terpenoids than mature tissue8. Generally, mature plant tissues of are not preferred for DNA extraction mainly due to the presence of high polysaccharides, polyphenols and other secondary metabolites9,10.
DNA yield and quality: The extraction method had a significant effect on DNA yield (Table 3). The DNA extracted by the SDS method was significantly higher than those obtained by C-TAB and plant DNAzol® kit.
 | |
| Fig. 1(a-d): | Color of the DNA pellet of G. xanthochymus extracted from different protocols, (a) Plant DNAzol® kit, (b) SDS buffer, (c) C-TAB buffer and (d) After purification of LiCl2 |
| Table 3: | Comparison of DNA quality (260/280) obtained by three extraction methods and purification |
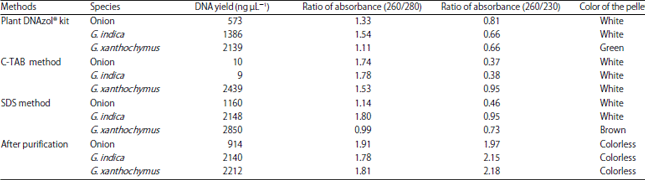 | |
The SDS method was most suitable for G. indica and onion and the color of the pellets were white. The DNA of G. xanthochymus extracted by SDS and kit method showed colored pellets containing high amount of polyphenol, polysaccharides and protein contamination (Fig. 1a, b). The color of the pellet was because of leaf material. Hence, C-TAB method was most suitable for G. xanthochymus as the purity of DNA was higher than compared to other methods after purification with LiCl2 (Fig. 1c, d). The phenolic compounds get oxidized and irreversibly bind to nucleic acids and various proteins at the time of tissue homogenization11,12. High content of polysaccharides make amplification of DNA difficult in the PCR due to its viscous nature, inhibiting the activity of Taq polymerase and also affect the activity of restriction enzymes13. The G. xanthochymus had very sturdy and hard leaves in earlier stage but G. indica had tender leaves and gave white color DNA pellet.
In both the protocols, we have tried to remove the cellular membrane with SDS and C-TAB buffer. Both these reagents are mild detergents and hence surfactants. The SDS is also chaotropic in nature which aids in disrupting proteins. The quantity and quality of DNA extracted by three different methods, significantly varied among the plant species tested (Fig. 2). The DNA yield by the SDS method was significantly higher than those obtained by the C-TAB method and plant DNAzol® kit.
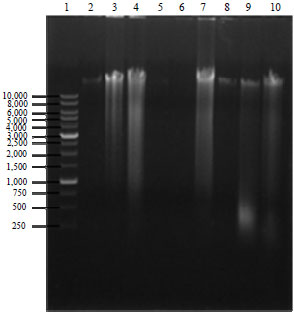 | |
| Fig. 2: | DNA extracted from onion, G. xanthochymus and G. indica by different methods on 0.8% agarose gel. 1: 1 kb ladder, Lane 2-4: DNA extracted from onion, G. indica and G. xanthochymus, respectively by plant DNAzol® kit, Lane 5-7: DNA extracted from onion, G. indica and G. xanthochymus by C-TAB method, Lane 8-10: DNA extracted from onion, G. indica and G. xanthochymus by SDS method |
In both the protocol development, we have tried to exploit the properties of triton X in the second purification step to remove lipid and protein component of the cellular membranes. Triton X helps in decreasing the surface tension and along with that it helps in solubilization of non-polar entities. High concentration of LiCl2 (0.5 M) in presence of ethanol helps in salting out of DNA.
In this study, higher yield of DNA was obtained from G. indica using the SDS method, probably because the young seedlings contained less secondary metabolite and for G. xanthochymus, C-TAB was the most preferable method. The lowest DNA yield was obtained by the method reported by Doyle and Doyle6 from G. indica and onion. The DNA extracted using these three methods were visualized on 0.8% agarose gel presented in Fig. 2. As compared to the kits SDS method showed relatively light smears, indicating less DNA degradation.
PCR amplification of RAPD, ISSR and rbcL primers: The PCR is considered one of the most efficient modes of indexing14. High quality of DNA is the most essential factor for a successful amplification by PCR15.
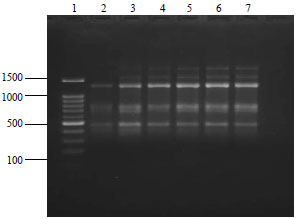 | |
| Fig. 3: | RAPD fingerprints for G. xanthochymus samples extracted and purified using modified C-TAB protocol with RP2 primer, loading sequence: Lane 1: 100 bp ladder, Lane 2-7: G. xanthochymus populations |
Isolation of highly purified plant DNA is difficult, particularly from plants like Garcinia with high polyphenolic compounds16 and these phenolic compounds hamper biochemical and molecular studies in Garcinia. Due to difficulties in isolation of pure DNA these polyphenols get oxidised resulting in quinonic compounds that cause browning of DNA preparations17.
The RAPD, ISSR and rbcL primers were successful in the amplification of DNA from G. xanthochymus, extracted by C-TAB method and G. indica extracted by the SDS method, indicating that these two methods were good and were giving sufficient quality of DNA for PCR amplification. On amplification using RAPD primer the isolated DNA showed high intensity bands (Fig. 3). The method is simple and can be worked out using in house compounds. We have also tried modified protocols of C-TAB and SDS with LiCl2 treatment for purification of DNA. The purity of the extracted DNA was excellent as evident by A260/A280 ratio ranging from 1.78-1.91 and A260/A230 ratio was above 2, suggesting that the preparations were sufficiently free of proteins and polyphenolics or polysaccharide compounds (Table 3). Clear banding patterns were observed in the RAPD (Fig. 3, 4) and ISSR study (Fig. 5). The extracted DNA was also amenable for amplification of plant barcode genes as evident in Fig. 6. The successfully amplified barcode gene sequences were purified, sequenced and submitted to GenBank (Accession No. KX291003 and KX522652). Figure 6 shows amplification of rbcL gene in Garcinia species. Impure DNA samples shows presence of smearing on the gel after amplification. Such impure DNA may be the cause for generating mix data and poly tail stretches of nucleotides during sequencing. Fresh and young leaf materials are the first choice to obtain good quality DNA.
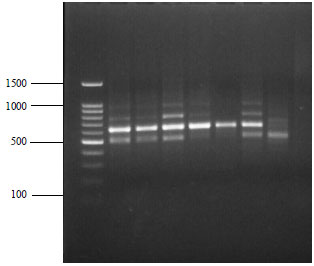 | |
| Fig. 4: | RAPD fingerprints for G. indica samples extracted and purified using modified SDS protocol with RP4 primer, loading sequence: Lane 1: 100 bp ladder, Lane 2-8: G. indica populations |
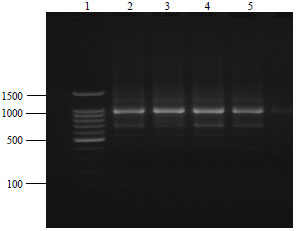 | |
| Fig. 5: | ISSR fingerprints for G. xanthochymus samples extracted and purified using modified C-TAB protocol with primer M10, loading sequence: Lane 1: 100 bp ladder, Lane 2-5: G. xanthochymus populations |
As, mature leaves contain higher quantities of polyphenols and polysaccharides, which make it very difficult to isolate DNA of good quality. There was no DNA fragmentation due to shearing of DNA during extraction procedure was seen in any of samples and the results were reproducible. The absence of smear further substantiates the high purity of extracted DNA. It has been reported previously that shearing of DNA during extraction can directly or indirectly interfere with the enzymatic reactions in different molecular studies like DNA barcoding. Sometimes such low quality DNA would generate mix data and there is a nucleotide poly tail stretch in between the sequences.
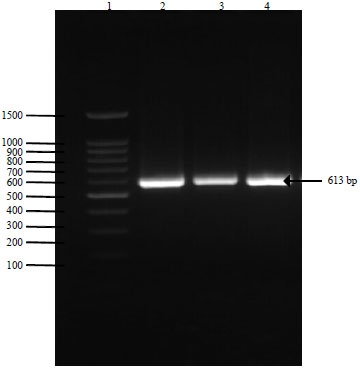 | |
| Fig. 6: | Amplification of rbcL barcode primer on Garcinia species, loading sequence: Lane 1: 100 bp ladder, Lane 2 and 3: G. indica, Lane 4: G. xanthochymus, respectively |
So, it may be an issue of DNA template. No inhibition of Taq DNA polymerase activity was observed in this study. Repeated chloroform:isoamyl alcohol treatment ensured removal of chlorophyll, pigments and dyes. This protocol could also be useful in other plant species with high polyphenols and polysaccharides.
DISCUSSION
The importance of Garcinia species as fruit trees is well recognized although its horticultural potential remains little exploited or developed. The G. indica, G. gummi-gutta and G. xanthochymus are the most important fruit trees in Western Ghats. The PCR is considered one of the most efficient modes of indexing14 and high quality of DNA is one of the essential factors for successful amplification by PCR15. Isolation of highly purified DNA is difficult from tree species, particularly from plants like Garcinia with high polyphenolic compounds18. Phenolic compounds are ubiquitous in leaves of Garcinia species. At the same time, the phenolic compounds hamper biochemical and molecular studies in Garcinia species, as isolation of pure DNA becomes difficult. Polyphenols often damage the DNA and also render the DNA inaccessible for some enzymes19. Protocol for extracting DNA appropriate PCR may include different filtration process and treatment with various specific and hazardous chemicals and reagents. Therefore, it was necessary to establish an inexpensive and efficient protocol for Garcinia species.
The two new protocols developed and optimized in this study yielded good quality DNA with a ratio of absorbance at A260/280 of 1.78 and 1.81 for G. indica and G. xanthochymus, respectively. This indicates that the DNA fraction was free of protein impurities and could be used for further applications. The presence of polysaccharide in the DNA sample inhibits enzyme activity20, but the DNA extracted using SDS and C-TAB was free of polysaccharides. This ratio is used as a secondary measure of nucleic acid purity. Successful amplification of RAPD, ISSR and rbcL gene with the DNA extracted by these methods further validates our experiment. Both the protocols yielded good quantity of DNA. The commercial kit used for DNA extraction protocol was not highly efficient for tree species containing phenolic compounds and hence not practically viable to be used by researchers. In this study we have developed two economically viable, efficient and rapid DNA isolation methods, which had more efficiency than commercial kits.
The current method developed to improve isolation of genomic DNA from leaf tissue of Garcinia plants species is a marked improvement over the earlier reported methods. Using this improved method, high quality of DNA was isolated from species of Garcinia that was amplified for RAPD, ISSR and barcode gene despite of high phenolics and polysaccharides. The optimized protocols are robust and efficient so as to make whole genome sequencing possible from Garcinia species for advanced bioinformatics investigations.
CONCLUSION
| • | It is evident that SDS is the best method in terms of quantity, quality and suitability for amplification of DNA from plants containing high content of phenolics and polysaccharides |
| • | The DNA obtained was of high quality and hence, amenable to RAPD, ISSR and barcoding analysis |
| • | Further, the materials and methods used were inexpensive and environmental friendly providing accurate results in a short duration |
SIGNIFICANCE STATEMENT
The methods optimized are able to yield a high quality DNA in minimum number of purification steps. This methods would be beneficial to other researchers while working with plants containing high phenolic and polysaccharides.
ACKNOWLEDGMENT
This study was supported by the Science and Engineering Research Board, Department of Science and Technology, New Delhi, Government of India, SERB sanction order No. and date: SB/YS/LS-25/2014.
REFERENCES
- Abraham, Z., S.K. Malik, G.E. Rao, S.L. Narayanan and S. Biju, 2006. Collection and characterisation of malabar tamarind [Garcinia cambogia (Gaertn.) Desr.]. Genet. Resour. Crop Evol., 53: 401-406.
CrossRefDirect Link - Ramage, C.M., L. Sando, C.P. Peace, B.J. Carroll and R.A. Drew, 2004. Genetic diversity revealed in the apomictic fruit species Garcinia mangostana L. (mangosteen). Euphytica, 136: 1-10.
CrossRefDirect Link - Pangsuban, S., N. Bamroongrugsa, K. Kanchanapoom and C. Nualsri, 2007. An evaluation of the sexual system of Garcinia atroviridis L. (Clusiaceae), based on reproductive features. Songklanakarin J. Sci. Technol., 29: 1457-1468.
Direct Link - Siatka, T. and M. Kasparova, 2010. Seasonal variation in total phenolic and flavonoid contents and DPPH scavenging activity of Bellis perennis L. flowers. Molecular, 15: 9450-9461.
CrossRefDirect Link - Dellaporta, S.L., J. Wood and J.B. Hicks, 1983. A plant DNA minipreparation: Version II. Plant Mol. Biol. Rep., 1: 19-21.
CrossRefDirect Link - Dabo, S.M., E.D. Mitchell and U. Melcher, 1993. A method for the isolation of nuclear DNA from cotton (Gossypium) leaves. Anal. Biochem., 210: 34-38.
CrossRefDirect Link - Zhang, J. and J.M. Stewart, 2000. Economical and rapid method for extracting cotton genomic DNA. J. Cotton Sci., 4: 193-201.
Direct Link - Loomis, W.D., 1974. Overcoming problems of phenolics and quinones in the isolation of plant enzymes and organelles. Methods Enzymol., 31: 528-544.
CrossRefDirect Link - Aljanabi, S.M., L. Forget and A. Dookun, 1999. An improved and rapid protocol for the isolation of polysaccharide- and polyphenol-free sugarcane DNA. Plant Mol. Biol. Rep., 17: 1-8.
CrossRefDirect Link - Porebski, S., L.G. Bailey and B.R. Baum, 1997. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep., 15: 8-15.
CrossRefDirect Link - Mullis, K.B. and F.A. Faloona, 1987. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol., 155: 335-350.
CrossRefPubMedDirect Link - Ahmed, I., M. Islam, W. Arshad, A. Mannan, W. Ahmad and B. Mirza, 2009. High-quality plant DNA extraction for PCR: An easy approach. J. Applied Genet., 50: 105-107.
CrossRefPubMedDirect Link - Sahasrabudhe, A. and M. Deodhar, 2010. Standardization of DNA extraction and optimization of RAPD-PCR conditions in Garcinia indica. Int. J. Bot., 6: 293-298.
CrossRefDirect Link - Johari, S. and S. Majumder, 2015. An efficient DNA extraction protocol for successful PCR detection of Banana bunchy top virus from banana leaves. Asian J. Biotechnol., 7: 80-87.
CrossRefDirect Link - Couch, J.A. and P.J. Fritz, 1990. Isolation of DNA from plants high in polyphenolics. Plant Mol. Biol. Rep., 8: 8-12.
CrossRefDirect Link - Fang, G., S. Hammar and R. Grumet, 1992. A quick and inexpensive method for removing polysaccharides from plant genomic DNA. BioTechniques, 13: 52-56.
PubMed