
D. McGrowder
Department of Pathology, University of the West Indies, Kingston 7, Jamaica
LiveDNA: 1876.2190
ORCID: 0000-0001-8018-7722
P.D. Brown
Department of Basic Medical Sciences (Biochemistry Section), University of the West Indies, Kingston 7, Jamaica
Asian Journal of Biochemistry
Year: 2007 | Volume: 2 | Issue: 1 | Page No.: 1-18







ABSTRACT
Since the discovery of Nitric Oxide (NO) as an important mediator of vasoregulation, NO has been found to be a diverse biological molecule. The continual release of NO is important for resting vascular tone and more recently, the role of NO in skeletal muscle glucose metabolism and contractile function has been increasing recognized. There is overwhelming evidence that endogenous NO is a critical mediator of insulin- and/or contraction-stimulated glucose transport. However, investigations of the role of exogenous NO donors in glucose metabolism in skeletal muscles have provided conflicting responses. There is evidence that insulin stimulation of glucose uptake in skeletal muscles is NO-dependent and a defect in insulin-induced vasodilation play a role in the pathogenesis of insulin resistance in hypertension, obesity and type 2 diabetes mellitus. The mechanism of insulin resistance by NO may possibly involve key components of the insulin signal transduction pathway. In addition, it is suggested that NO is involved in contraction-stimulated glucose uptake through a mechanism that is distinct from the insulin signal transduction pathway. In light of the conflicting reports and unclear mechanisms of NO in basal, insulin- and contraction-stimulated glucose uptake, this review examines recent developments and explore whether the possible role of NO in regulation of skeletal muscle and adipose tissue uptake may have important clinical significance.
PDF Abstract XML References Citation
How to cite this article
D. McGrowder and P.D. Brown, 2007. Effects of Nitric Oxide on Glucose Transport: in vivo and in vitro Studies. Asian Journal of Biochemistry, 2: 1-18.
DOI: 10.3923/ajb.2007.1.18
URL: https://scialert.net/abstract/?doi=ajb.2007.1.18
DOI: 10.3923/ajb.2007.1.18
URL: https://scialert.net/abstract/?doi=ajb.2007.1.18
INTRODUCTION
Nitric Oxide Synthases-regulation
Over the past two decades there has been increasing evidence of the many and diverse biological functions of endogenous Nitric Oxide (NO) in the cardiovascular, nervous and immune systems. Nitric oxide is produced in a variety of tissues from the amino acid L-arginine through the activation of different isoforms of NO Synthase (NOS) (Moncada and Higgs, 1991). Three members of the NOS family have been identified and they are neuronal NOS (nNOS), endothelial NOS (eNOS) and inducible NOS (iNOS) (Kobzik et al., 1994). Neuronal NOS is the most abundant isoform found in skeletal muscle and is located preferentially at neuromuscular junctions (Kusner and Kaminski, 1996). Endothelial NOS is present in the vascular endothelium and in skeletal muscles. Both nNOS and eNOS synthesize small amounts of NO and require activation by Ca2+-calmodulin, making them sensitive to agents and processes that increase intracellular calcium (Abu-Soud and Stuehr, 1993). The NO generated diffuses to neighboring target cells where it acts primarily through the activation of soluble guanylate cyclase (sGC) which catalyzes the formation of cyclic guanosine monophosphate (cGMP) from guanosine triphosphate (GTP). This cause a cellular response through a reduction in intracellular calcium levels (Waldman and Murad, 1987). Inducible NOS is present in macrophages and is localized at the sarcolemma of skeletal muscle cells where its activity varies between species and is dependent on the disease state (Kobzik et al., 1995). The enzyme is induced under certain pathological conditions and the high NO output from iNOS results in the concentrations of NO reaching toxic levels (Moncada et al., 1991).
Studies have showed that NO mediates exercise-stimulated glucose transport in isolated skeletal muscle (Balon and Nadler, 1997) and NO released from skeletal muscle plays a key role in the control of metabolic homeostasis in both animals (Duplain et al., 2001) and humans (Scherrer and Sartori, 2000). Defective eNOS-driven NO synthesis causes insulin resistance in experimental animals (Shankar et al., 2000) and characterizes insulin-resistant states in humans (Sartori and Scherrer, 1999). A novel role for NO in the regulation of adipose tissue function has emerged from studies which have provided evidence for the expression of NO synthase type II and III isoforms in adipose tissue.
On the other hand, the induction of iNOS and NO overproduction may alter glucose metabolism in muscle and adipose tissue (Bedard et al., 1997). The induction of the expression of the macrophage-type iNOS in skeletal muscle cells by cytokines and endotoxin increases basal glucose uptake. However, the ability of insulin to stimulate glucose uptake was impaired suggesting that NO overproduction may be involved in the cytokine-induced insulin resistance in skeletal muscle (Bedard et al., 1997). In myocytes, cytokines and lipopolysaccharides induce iNOS expression and augment basal glucose transport, but abolish insulin-stimulated glucose transport, an effect that was reversed by the non-specific NOS inhibitor NG-nitro-L-arginine methyl ester (L-NAME) (Kapur et al., 1997). Conversely, in adipose cells iNOS inhibition did not attenuate cytokine-induced stimulation of basal glucose uptake suggesting that the regulation of glucose metabolism in adipose cells is independent of NO (Pilon et al., 2000).
Research on the effect of NO donors on glucose metabolism in skeletal muscle, cardiac muscle, smooth muscle and adipose tissue has grown rapidly in the last decade. Most research has focused on skeletal muscle and the role of NO in the modulation of skeletal muscle glucose transport remains controversial. This review focuses on recent evidences in the literature on the role of both exogenous and endogenous on glucose metabolism, specifically glucose uptake in peripheral and myocardial tissues. In addition we examine proposed mechanism by which NO modulates basal, insulin- and contraction-stimulated glucose uptake which involve signaling pathways.
Nitric Oxide Mediation of the Vasodilatory Action of Insulin
Nitric oxide has an important role in the control of basal and insulin-stimulated blood flow in humans (Boczkowski et al., 1994). Vallance and colleagues investigated the effects of endothelium-derived NO on peripheral arteriolar tone in man and found that arterial infusion of the NOS inhibitor NG-monomethyl-L-arginine (L-NMMA) caused a 50% fall in basal blood flow and attenuated the dilator response of acetylcholine (an endothelium-dependent inhibitor). This response was not shown with glyceryl trinitrate, demonstrating that endothelium-derived NO contributes to basal and stimulated regional blood flow in humans (Vallance et al., 1989).
Skeletal muscle is the primary target tissue for insulin stimulation of glucose transport, a regulatory mechanism vital for glucose homeostasis. Insulin increases glucose transport in this tissue mainly by activating the translocation of glucose transporter 4 (GLUT-4) from an occluded intracellular tubulo-vesicular reservoir to the cell surface (Khan, 1992). There is evidence that insulin enhances skeletal muscle glucose disposal by vasodilating the muscle vasculature (Baron, 1994). The increase in muscle perfusion (Baron et al., 1996) is thought to increase the delivery of glucose to muscle cells (Baron et al., 1995). Baron and colleagues suggests that approximately thirty percent (~ 30%) of insulin’s effects on glucose uptake can be accounted for by increases in muscle perfusion (Baron et al., 1995). However, there is evidence that the enhancing effect of insulin on the musculature is mediated by the release of NO from the endothelium (Scherrer et al., 1994). Previous studies have shown that abrogation of NO release by L-NMMA prevented the action of insulin to increase blood flow to skeletal muscle (Steinberg et al., 1994). In addition, the removal of the endothelium or NOS inhibition prevents the vasodilating action of insulin on skeletal muscle arterioles (Chen and Messina, 1996).
Insulin increases blood flow and glucose delivery in skeletal muscle of both humans (Baron, 1994) and rats (Pitre et al., 1996) and there is further evidence that insulin stimulation of glucose uptake in skeletal muscles is NO-dependent. Baron and his colleagues reported that insulin administered by continuous intravenous infusion for hours, increases leg blood flow (Baron et al., 1988; Baron and Clark, 1997). In humans, induced acute hyperinsulinaemia caused a significant increase in urinary excretion of nitrite (NO2−)/nitrate (NO3−) together with a significant decrease in blood pressure, supporting the concept that NO may mediate the vasodilatory action of insulin in humans (Tsukahara et al., 1997).
The acute administration of the NOS inhibitors L-NMMA or L-NAME results in the development of marked insulin resistance, hypertension and/or hyperglycaemia (Shankar et al., 1998). The blockade of NOS decreased blood flow to skeletal muscle and impairs insulin-mediated glucose disposal during a hyperinsulinaemic-euglycemic clamp in vivo (Roy et al., 1998) which is in agreement with a previous study in human subjects (Steinberg et al., 1994). In another study, Butler and colleagues found that L-NMMA infusion increases rather than decreases whole body glucose transport in 16 healthy subjects and that the NOS inhibitor also increased rather than decreased calf blood flow with minimal change in the blood pressure (Butler et al., 1998). The hemodynamic responses of L-NMMA were opposite from those observed in previous studies, including those in which it caused insulin resistance (Baron et al., 1995). In contrast to the effects of NOS inhibition in vivo, NOS inhibitors fail to affect insulin-stimulated glucose transport in isolated muscles incubated using in vitro preparation (Young et al., 1997a, b). The authors suggest that hemodynamic factors are needed to fully amplify the increase in insulin-stimulated glucose transport in skeletal muscle. In addition, Roy et al. (1998) found that NOS inhibition in vitro had no effect on insulin-stimulated glucose transport .
Nitric Oxide Donors and Glucose Transport
The role of NO in the modulation of skeletal muscle glucose transport and metabolism is conflicting. There are reports that SNP at concentrations of 1-25 mM increases glucose transport induced by submaximal concentrations of insulin (Balon and Nadler, 1997; Etgen et al., 1997) and glucose oxidation (Young and Leighton, 1998) in isolated muscle preparations. With higher concentrations of SNP, glucose uptake decreased (Balon and Nadler, 1997). However in another study by Kapur and colleagues, 0.1-1 mM of SNP and 10-100 M GEA 5024 [1,2,3,4-oxatriazolium, 5-amino-3-(3-chloro-2-methylphenyl)-chloride] had an opposite effect (Kapur et al., 1997). SNP enhanced glucose uptake by increasing GLUT4 expression on the cell surface of isolated rat epitrochlearis muscle (Etgen et al., 1997). However, adequate controls for SNP at these concentrations were not performed and more specific NO donors were not tested. In addition, differences between the studies may be due to the dose of the NO donor administered and the experimental model used.
In a more recent study by McGrowder and colleagues the NO donors S-nitrosoglutathione (GSNO) and S-nitroso-N-acetylpenicillamine at 0.2-1.0 mM stimulated basal and insulin-stimulated glucose uptake, while concentrations of 10 mM and 20 mM inhibited basal glucose uptake in isolated muscle strips of normoglycemic and streptozotocin-induced diabetic rats (McGrowder et al., 2006) (Fig. 1 and 2). The magnitude of the reduction of glucose uptake using GSNO and SNAP was greater in the diabetic rats. S-nitroso-N-acetylpenicillamine was found to have a more significant effect on basal and insulin-stimulated glucose uptake than GSNO in the normoglycaemic rats (McGrowder et al., 2006) and prior investigations by the same authors demonstrated that SNAP generates more NO than GSNO (McGrowder et al., 1999, 2001).
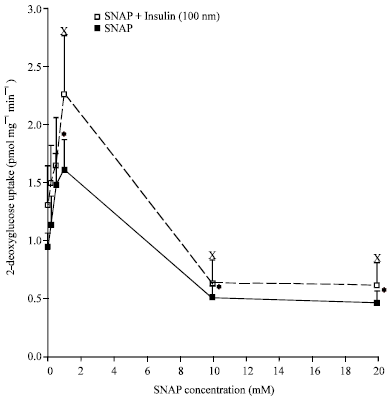 | |
| Fig. 1: | Graph showing the effects of SNAP on basal and insulin-stimulated glucose uptake in skeletal muscles of STZ-induced diabetic rats. Statistical significant differences are indicated by *p<0.05 vs uptake in basal, Xp<0.05 vs uptake with insulin (Reprinted with permission from McGrowder et al., 2006) |
Insulin Resistance and Nitric Oxide
Insulin increases skeletal muscle blood flow by a Nitric Oxide (NO)-dependent mechanism and impairment of this mechanism may contribute to the insulin resistance that is characterized by reduced endothelial production of NO, an attenuated effect of insulin on skeletal muscle blood flow and resistance to insulin-mediated glucose uptake (Potenza et al., 2005). Nitric oxide donors may possess some potential value in the treatment of insulin resistance in animals. In a study by Oshida and colleagues, SNP was found to improve insulin resistance induced by high-fructose feeding and restore insulin sensitivity during sequential hyperinsulinemic euglycemic clamp studies in insulin-resistant high-fructose-fed rats and control rats (Oshida et al., 2000). Another NO donor, NO-aspirin restored insulin sensitivity in a mouse model of insulin resistance associated with defective endothelial synthesis (Cook et al., 2001).
Essential hypertension, like obesity and type 2 diabetes mellitus diabetes, is a condition associated with reduced sensitivity of skeletal muscle tissues to the action of insulin on glucose uptake (Natali et al., 1991). In all these conditions, the limb vasodilation that follows systemic insulin infusion also appears to be impaired (Laakso et al., 1989; Baron et al., 1993) although this finding is not in agreement with other studies (Egan and Stepniakowski, 1994; Dela et al., 1995). Consequently a defect in insulin-induced vasodilation has been proposed to play a role in the pathogenesis of insulin resistance in these conditions (Baron et al., 1995). Insulin resistance is characterized by reduced endothelial production of NO, an attenuated effect of insulin on skeletal muscle blood flow and resistance to insulin-mediated glucose uptake (Meneilly et al., 2001).
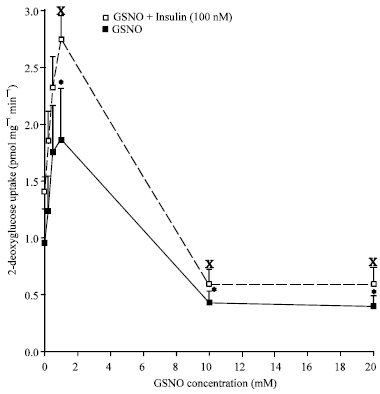 | |
| Fig. 2: | Graph showing the effects of GSNO on basal and insulin-stimulated glucose uptake in skeletal muscles of STZ-induced diabetic rats. Statistical significant differences are indicated by *p<0.05 vs uptake in basal, Xp<0.05 vs uptake with insulin (Reprinted with permission from McGrowder et al., 2006) |
The role of insulin-stimulated blood flow as a determinant of glucose uptake has been studies mainly be increasing or decreasing blood flow with various vasoactive agents and then determining the impact of such an intervention on glucose uptake. Systemic intravenous or local intra-arterial infusion of the endothelium-independent vasodilator SNP in normal and insulin-resistant humans has been used to augment muscle blood flow during euglycemic-hyperinsulinemic clamp studies. In a study by Meneilly et al. (2000) systemic infusion of SNP augmented blood flow but did not increase insulin-mediated glucose disposal in young and old subjects. Others found that in overweight male patients with essential hypertension, increasing forearm perfusion with SNP does not attenuate insulin forearm tissues (Natali et al., 1998) and that vasodilation induced by methacholine but not SNP increased glucose uptake in the forearm of hypertensive patients (Sarabi et al., 1999). The authors concluded that an increase in forearm blood flow does not necessarily improve glucose uptake in the forearm during the fasting state (Sarabi et al., 1999). These observations suggest that the acute delivery of large intravascular doses of NO into the skeletal vasculature may be inhibitory rather than stimulatory and therefore may not be an appropriate way to augment muscle glucose disposal in normal and insulin-resistant subjects. L-arginine is a precursor for NO and both in vivo and in vitro studies have demonstrated that L-arginine can augment vascular dilation under certain conditions (Jun and Wennmalm, 1994). In a study by Piatti and colleagues, long-term oral administration of L-arginine significantly improved but not completely normalized peripheral and hepatic insulin sensitivity in type 2 diabetic patients (Piatti et al., 2001).
Mechanism of Action of Nitric Oxide Donors
There is evidence that a close link exists between iNOS and insulin resistance and most if not all inducers of insulin resistance increase iNOS expression. These inducers of insulin resistance include obesity (Elizalde et al., 2000), hyperglycaemia (Ceriello et al., 2002), tumour necrosis factor-α (TNF-α) and endotoxin. Inducible NOS mediates the impaired insulin-stimulated glucose uptake by treatment with TNF-α and lipolysaccharide in cultured skeletal muscle cells and its expression is elevated in skeletal muscle of patients with type 2 diabetes mellitus (Torres et al., 2004) and high fat diet-induced mice (Perreault and Marette, 2001). Sugita and colleagues demonstrated that iNOS inhibitor prevents lipopolysaccharide-induced insulin resistance in rats (Sugita et al., 2005). In another study, it was demonstrated that genetic disruption of iNOS protects against obesity-linked insulin resistance, preventing impairments in phosphatidylinositol 3-kinase (PI3K) and protein kinase B/Akt activation by insulin in skeletal muscle (Perreault and Marette, 2001).
S-nitrosation and S-nitrosylation of Components in Signaling Pathway
The molecular mechanisms by which iNOS mediates insulin resistance is still being investigated and it is suggested that components of the insulin signal transduction pathways are affected. This pathway plays a central role in metabolic actions of insulin, including stimulation of glucose uptake, synthesis of glycogen and protein and inhibition of gluconeogenesis (Agati et al., 1998). Insulin receptor substrate (IRS)-1 is a key molecule in insulin-signaling that transduces a signal from insulin receptor (IR) to phosphatidylinositol-3-kinase (PI3K) (Saltiel and Khan, 2001). Inducible NOS induction (Bedard et al., 1997) and two classes of NO donors (Bedard et al., 1998) inhibited insulin-stimulated glucose transport in isolated soleus and EDL muscles and cultured L6 muscle cells. Further investigations showed that exposure to GSNO or iNOS transfection reduced IRS-1 protein expression via proteasome-mediated degradation without altering the mRNA level in cultured skeletal muscles (Sugita et al., 2005). The effect of GSNO on IRS-1 expression was cGMP-independent and GSNO and insulin seem to exert the effects on IRS-1 via different pathways (Sugita et al., 2005). Previous studies have demonstrated that the activation of PI3K and mammalian targets of rapamycin (mTOR) is required for IRS-1 degradation by insulin in cultured muscle cells (Greene et al., 2003). Unlike insulin, GSNO does not require PI3K and mTOR activity to reduce IRS-1 expression (Sugita et al., 2005) and it may probably involve intracellular nitrosative protein modification such as S-nitrosylation (Eu et al., 2000).
Protein S-nitrosylation involving attachment of nitrosonium ion (NO+) to cysteine sulfhydryls, has emerged recently as a prototype of cGMP independent, redox-dependent post-translational modifications (Stamler et al., 2001) which mediates a number of actions of the NO group in various biological processes (Gow et al., 2002; Hess et al., 2001). Protein kinase B/Akt is a serine/threonine protein kinase which plays a central role in the metabolic actions of insulin (Jiang et al., 2003), including glucose transport and is impaired in rodent models of insulin resistance and patients with type 2 diabetes (Carvalho et al., 2000; Shao et al., 2000).
Insulin activates PI3K-Akt/PKB via IRS-1 and -2 in skeletal muscle and adipocytes (White and Khan, 1994). In a study by Yasukawa and colleagues, NO released from GSNO and SNAP were found to induced S-nitrosylation and inactivation of Akt/PKB in vitro and in intact cells. The inhibitory effects of NO were independent of PI3K and cGMP and S-nitrosylation of Akt/PKB was increased in skeletal muscle of diabetic (db/db) mice compared with wild type mice (Yasukawa et al., 2005). The authors suggests that S-nitrosylation-mediated inactivation may contribute to the diabetic complications via selective impairment in insulin signaling and the pathogenesis of iNOS and/or oxidative stress involved in insulin resistance (Yasukawa et al., 2005). Another recent study found that NO donated by GSNO induced in vitro and in vivo S-nitrosation of the IR β subunit (IRβ) and protein kinase B/Akt and reduce their kinase activity in muscle. Insulin receptor-1β was also rapidly S-nitrosated and its expression was reduced after chronic GSNO treatment (Carvalho-Filho et al., 2005). In addition, in two distinct models of insulin resistance associated with enhanced iNOS expression (diet-induced obesity and the ob/ob diabetic mice) there was enhanced S-nitrosation of IRβ/IRS-1 and Akt in muscle (Carvalho-Filho et al., 2005). These observations suggests that NO donated from its donors and an increase in iNOS expression induce down-regulation of insulin signaling by altering different components of the insulin signal transduction pathway. This provides a mechanism by which iNOS expression is induced in situations associated with insulin resistance.
Contraction-stimulated Glucose Uptake
NO-dependent Mechanism Involving Glut-4
During the past ten years, it has been proposed that NO mediates exercise-stimulated glucose transport in skeletal muscle (Balon and Nadler, 1997; Roberts et al., 1997). During exercise, skeletal muscle glucose uptake and blood flow substantially increase. Studies in animals assessing the role of NO in exercise/contraction-stimulated glucose uptake have resulted in contrasting conclusions (Balon and Nadler, 1997; Etgen et al., 1997; Roberts et al., 1997). In one of these studies, hind-limb muscles were contracted in situ via electrical stimulation of the sciatic nerve and the extensor digitorum longus (ELD) muscles were isolated and used for the measurement of glucose transport in the presence or absence of L-NMMA (Balon and Nadler, 1997). Under these conditions, L-NMMA was shown to block contraction-stimulated glucose transport. Similar results were also observed in female Sprague-Dawley rats that were first exercised on a treadmill and given L-NAME in drinking water (Roberts et al., 1997). In contrast, in the study by Etgen and colleagues, there was normal activation of contraction-stimulated glucose transport when isolated rat epitrochlearis muscle preparations were treated with L-NMMA (Etgen et al., 1997). In addition, in a later study L-NMMA failed to affect exercise or contraction-stimulated 2-deoxyglucose uptake in hind-limb muscle contracted in vivo via electrical stimulation of the sciatic nerve, isolated EDL muscles or isolated soleus muscle from rats after they had performed running exercise for 1 h (Higaki et al., 2001). These observations suggest that there are differences between in vivo and in vitro muscle stimulation in animals.
Nitric oxide has been implicated in the putative mechanisms proposed for contraction-mediated glucose transport in humans (Bradley et al., 1999; Kingwell, 2002). The NO inhibitor, L-NMMA reduced leg glucose by 48% during cycle exercise in humans in the absence of any effect on leg blood flow or plasma insulin (Bradley et al., 1999). During exercise, glucose uptake can occur in the absence of insulin (Ploug et al., 1984). Under physiological conditions, however, both insulin and contraction are important synergistic mediators of glucose uptake during exercise such that for any given level of contractile activity, insulin further increases glucose uptake (De Fronzo et al., 1981; Hespel et al., 1996). Studies have showed that individuals with type 2 diabetes (Zierath et al., 1996) and insulin-resistant obese Zucker rats (King et al., 1992) have impaired insulin-stimulated GLUT-4 translocation; however, exercised-stimulated GLUT-4 translocation is normal (King et al., 1993). Furthermore, despite deficits in insulin-mediated GLUT-4 translocation, skeletal muscle glucose utilization during exercise is normal (Minuk et al., 1981; Martin et al., 1995) or supra-normal in individuals with type 2 diabetes (Colberg et al., 1996). In a recent study, NO inhibition during exercise decreased glucose uptake in individuals with type 2 diabetes than in control subjects indicating that NO may be a key mediator of the majority of glucose extraction by skeletal muscle during exercise in individuals with type 2 diabetes (Kingwell et al., 2002).
Although exercise and insulin both significantly increased glucose transport, L-NAME had no effect on insulin-stimulated glucose transport but incompletely blocked both GLUT-4 translocation and exercise-stimulated glucose transport, indicating that NO is involved in the contraction-stimulated glucose transport signal transduction mechanism Roberts et al., 1997). Coderre and colleagues have isolated an intracellular exercise-sensitive pool of GLUT-4 transporters and suggested that this GLUT-4 protein may be associated with glycogen levels (Coderre et al., 1995). In addition, GLUT-4 concentration is a primary determinant of post-exercise muscle glycogen storage (McKoy et al., 1996). Consequently, in response to exercise, both GLUT-4 translocation and subsequent exercise-induced transport may depend on muscle glycogen utilization to release the associated GLUT-4 transporters. In a study by Landers and colleagues, NO activates p38 and c-Jun NH2-terminal kinase (JNK), both mitogen-activated protein (MAP) signaling kinases, suggesting that these molecules participate in NO signal transduction (Lander et al., 1996). Furthermore, Goodyear and colleagues demonstrated that p38 and JNK in skeletal muscle are increased in response to exercise but not to insulin, suggesting that these signal kinases are involved in cell signaling during exercise (Goodyear et al., 1996).
Insulin and Contraction-induced Pathways
AMP Kinase-induced Skeletal Muscle Glucose Uptake
There are at least two distinct signaling cascades that stimulate glucose transport in skeletal muscle. One pathway is stimulated by insulin and insulin-like growth factor-1. Phosphatidylinositol 3-kinase (PI3K) is necessary for activation of glucose transport by this mechanism (Cheatham et al., 1994; Clarke et al., 1994). The contraction or contraction/hypoxia pathway is PI3K independent (Goodyear et al., 1995; Lund et al., 1995). A study by Higaki and colleagues found that additive effect of SNP and insulin on skeletal muscle transport and that NO-stimulated transport was only partially inhibited by wortmannin, a PI3K inhibitor (Higaki et al., 2001). They suggest that this finding which is consistent with observations of other studies (Balon and Nadler, 1997; Etgen et al., 1997; Roy et al., 1998) demonstrate that NO signaling pathway mediates skeletal muscle glucose transport through an insulin-independent pathway. In addition, there is considerable evidence indicating that these effects occur via different mechanisms, because the two stimuli appear to recruit separate pools of GLUT-4 to the plasma membrane and the effect of insulin, but not exercise, is blocked by wortmannin (Baron et al., 1996; Kemp et al., 1999; Hardie and Carling, 1997). Wormannin has no effect on the stimulation of glucose transport by the AMPK activator, 5-amino-4-imidazolecarboxamide riboside (AICAR), indicating that AMP-activated protein kinase (AMPK) and insulin increase glucose transport in the skeletal muscle by different mechanisms (Hayashi et al., 1998).
The AMP-activated protein kinase (AMPK) cascade has been proposed to act as a metabolic master switch, regulating energy metabolism in response to changes in the energy charge of the cell (Winder and Hardie, 1999). It has been reported that activation of AMPK in muscle, using AICAR leads to increase in glucose transport (Merrill et al., 1997; Hayashi et al., 1998). The increase in glucose transport induced by AICAR is accompanied by increased translocation of GLUT4 to the plasma membrane (Kurth-Kraczek et al., 1999). To determine the dependence of AMPK on NO, a number of studies have activated AMPK in the presence or absence of NOS inhibitors. In cell culture, the combination of L-NMMA and AICAR resulted in the elimination of AICAR-stimulated glucose uptake, suggesting that AMPK increases glucose uptake by an NO-dependent pathway (Fryer et al., 2000). An inhibitor of guanylate cyclase also blocks the activation of glucose transport by AICAR demonstrating that the activation of AMPK in muscle cells stimulates glucose transport by activation of NOS coupled to downstream signaling components, including cyclic GMP (Fryer et al., 2000). Additional evidence is found by Morrow and colleagues, where activation of AMPK by AICR in human aortic endothelial cells resulted in an increase in NOS phosphorylation and NO production (Morrow et al., 2003). Furthermore, a significant study in the area was done by Higaki and colleagues who found that NO is not involved in the signaling pathway leading to contraction-stimulated glucose uptake in skeletal muscle and that SNP increases skeletal muscle glucose uptake through a mechanism that is distinct from the insulin- and contraction-signaling pathways. They proposed that the there is a third signaling pathway that enhances glucose uptake in skeletal muscle which is associated with an activation of the α1 catalytic subunit of AMPK (Higaki et al., 2001). Therefore, these in vitro studies provide strong evidence that AMPK acts via a NO pathway.
In vivo data are lacking and only a few studies have been done. In a recent study by Shearer and colleagues, AMPK activation simultaneously increased glucose and long-chain fatty acid (LCA) clearance and L-NAME impedes AMP-induced glucose uptake in skeletal muscle of male Sprague-Dawley rats (Shearer et al., 2004). In addition to determining the NO dependence of AMPK’s actions, this study demonstrates that both AMPK-induced glucose and LCA uptake are dependent on muscle fibre-type composition with muscles of a greater percentage of fast-twitch fibres more responsive to AMPK-stimulated glucose uptake than muscles comprised of more slow-twitch fibres (Shearer et al., 2004).
Myocardial Glucose Uptake and Nitric Oxide
In skeletal muscle of male Sprague-Dawley rats, NO affects myocardial utilization, exerting an inhibitory action on myocardial glucose uptake and metabolism (Depre et al., 1995). Exogenous NO, by means of its second messenger cGMP inhibits glucose uptake and utilization in ischemic as well as nonischemic isolated hearts (Depre et al., 1998) and in quiescent myocytes (Bergemann et al., 2001). Studies by Tada and colleagues showed that endogenous NO is likely to be responsible for a tonic inhibition of cardiac carbohydrate metabolism as shown by the marked elevation of glucose uptake under basal conditions, in isolated hearts from endothelial NOS knockout mice (Tada et al., 2000). There is evidence that SNP stimulates glucose uptake in cardiomyocytes via MAPK. During ischemia there is the activation of glucose uptake via translocation of GLUT-4 from an intracellular pool to the sarcolemma (Wheeler, 1988; Young et al., 1997a, b) and AMPK stimulates GLUT-4, glucose uptake and glycolysis (Marsin et al., 2000; Russell et al., 2004). In addition, SNP stimulates glucose uptake in cardiomyocytes via mitogen-activated protein kinase (Jensen et al., 2003). However, a recent study found that the activation of nitric oxide/guanylate cyclase pathway contributes to, but is not the sole mediator of AMPK-stimulation of glucose uptake and GLUT-4 translocation in heart muscle (Li et al., 2004).
Nitric oxide donors such as nitroglycerin (NTG) have been widely used in the treatment of angina as its vasodilatory effects are mediated through breakdown to form NO within the coronary and systemic blood vessels (Ignarro et al., 2002). Lei and colleagues found that in ischaemic myocardium, NO released by NTG inhibits glucose uptake and lactate production by a reduction of AMPK stimulation of GLUT-4 translocation, revealing a mechanism of metabolic modulation by NO donors (Lei et al., 2005). In addition, NO from NTG inhibits lactate production and the inhibition of NO synthesis stimulates glucose oxidation by means of a pyruvate dehydrogenase-independent mechanism. These mechanisms suggest that the therapeutic actions of NO donors in myocardial ischemia may be partially due to the metabolic effects of these agents (Lei et al., 2005). The results of this study by Lei and colleagues are in agreement with previous work in which reduced activation of AMPK, GLUT-4 translocation, glycolysis and lactate production in dogs receiving NO donors reflect a cause-effect relationship (Marsin et al., 2000; Russell et al., 2004).
Regulation of Nitric Oxide System in Adipose Tissue
A novel role for NO in the regulation of adipose tissue function has emerged in the last ten years (Anderson et al., 1999; Ribiere et al., 1996), providing evidence for the expression of NO synthase type II and III isoforms tissue. In a recent study by Tanaka and colleagues, eNOS was found to be expressed in murine 3T3-L1 adipocytes (Tanaka et al., 2003) and this result is consistent with previous findings in human adipocytes (Ribiere et al., 1996). The NO donor, SNP stimulated glucose uptake in 3T3-L1 adipocytes and insulin was found to induce GLUT 4 translocation and glucose uptake through phosphorylation of IRS-1 or Akt in 3T3-L1 adipocytes (Tanaka et al., 2003). Nitric oxide did not induce phosphorylation of IRS-1 and Akt during the stimulation of glucose uptake, indicating that the insulin receptor/Akt pathway is not involved in NO function (Tanaka et al., 2003). Based on these observations, the authors suggest it may appear that NO stimulate glucose uptake through an insulin-independent pathway in 3T3-L1 adipocytes (Higaki et al., 2001) and that GLUT-4 translocation is involved in the NO-stimulated mechanism (Tanaka et al., 2003).
NOS blockade was found to reduce insulin-mediated glucose uptake in both brown and white adipose tissues (Roy et al., 1998). The beneficial effects of NO formation on insulin-stimulated glucose uptake (Roy et al., 1998) are most likely explained by insulin-stimulated NO production in endothelial cells. NO may in turn facilitate glucose delivery into target organs muscle and adipose tissues by increasing tissue blood flow (Pilon et al., 2000). The AMPK cascade has been characterized in 3T3-L1 adipocytes (Salt et al., 2000). In contrast to skeletal muscle in which AMPK stimulation promotes glucose transport to provide ATP as a fuel, AMPK stimulation inhibits insulin-stimulated glucose transport in adipocytes, inhibiting triacylglycerol synthesis, to conserve ATP under conditions of cellular stress (Salt et al., 2000). Furthermore, AMP-activated kinase-mediated posttranslational phosphorylation inhibited iNOS activity and enhanced insulin sensitivity in muscle and adipose tissues (Pilon et al., 2004).
Glucose Uptake in Vascular Smooth Muscle and Nitric Oxide
Vascular smooth muscle cells (VSMCs) have been shown to express GLUT-4 and exhibit a significant insulin-responsive glucose uptake similar to that of skeletal muscle and adipose tissue (Kahn et al., 1995; Standley et al., 1995). It has been proposed that insulin-induced vasodilation is mediated, at least in part, by the stimulation of endothelial NO production, causing inhibition of contraction of the underlying VSMCs via activation of guanylate cyclase (GC) (Scherrer et al., 1994). A direct effect of insulin on cultured VSMCs has been observed in vitro, showing that these cells are targets of insulin action (Trovati and Anfossi, 2001). In addition, it has been found that insulin increases cGMP production by a NO-dependent mechanism (Trovati et al., 1995) and inducible NOS expression as well as cGMP generation via the PI3K pathway (Begum et al., 1998).
A recent study by Bergandi et al. (2003) found that insulin stimulates glucose transport and GLUT-4 translocation in human VSMCs owing to its ability to increase endogenous NO production via activation of NOS, as the NOS inhibitor L-NAME completely blocks both insulin-mediated effects. In addition, GSNO and SNP enhance both glucose transport and GLUT-4 translocation in human VSMCs, bypassing the inhibitory effect of L-NAME (Bergandi et al., 2003). The authors suggests that in human VSMCs, NO plays an important role in both insulin-dependent relaxation/vasodilation and glucose uptake (Bergandi et al., 2003).
The role of NO/cGMP signaling in glucose transport is different between smooth muscle and skeletal muscle. Fryer and colleagues found that elevation of glucose transport in rat skeletal muscle and mouse H-2k b muscle cells was completely blocked by inhibition of NOS only under those conditions that lead to increase AMPK activity. In addition, inhibitors of NOS have no effect on the elevation of glucose transport caused by stimuli such as insulin that do not activate AMPK (Fryer et al., 2000). Conversely, in human VSMC, the elevation in glucose transport is promoted by insulin via a NOS- and GC-dependent mechanism. The stimulating effects of insulin and cGMP on glucose transport require cGMP-dependent kinase (PKG) activation (Bergandi et al., 2003). This enzyme plays a central role in the insulin-dependent rapid stimulation of glucose transport in human VSMCs, which occurs via the ability of insulin to promote the translocation of intracellular vesicles containing GLUT-4 to the plasma membrane (Pfeifer et al., 1999). However the molecular mechanisms of cGMP signaling distal to PKG I are not yet completely understood and are at present being actively investigated.
Conclusions and Future Perspectives
Recent evidence suggests that NO is capable of stimulating glucose transport through GLUT-4 translocation in 3T3-L1 adipocytes, via a mechanism different from the insulin-signaling pathway and may involve AMPK. In ischemic myocardium, NTG inhibits glucose uptake and lactate production via reduction in AMPK stimulation of GLUT-4 translocation, revealing a mechanism of metabolic modulation and myocardial production activated by NO donors.
Defective eNOS driven NO synthesis causes insulin resistance, arterial hypertension and dyslipidemia in mice and characterizes insulin-resistance in humans. In addition, the stimulation of iNOS and NO overproduction in mice may also cause metabolic insulin resistance. Nitric oxide has been implicated as an important signaling molecule in the contraction-mediated glucose uptake pathway and may represent a novel strategy for blood glucose control. NO-mediated glucose transport may compensate for impaired insulin action and account for normal glucose uptake in individuals with type 2 diabetes during exercise. The NO pathway therefore may represent a potential therapeutic target in patients with type 2 diabetes.
The intracellular pathways that lead to contraction- and insulin-stimulated GLUT-4 translocation seem to be different, allowing a maximal effect when acting together. Insulin utilizes a PI3K-dependent mechanism, whereas the exercise signal may be initiated by calcium release from the sarcoplasmic reticulum or from autocrine- or paracrine-mediated activation of glucose transport. Furthermore, AMPK is activated by exercise in skeletal muscle which stimulates glucose transport by activation of NOS coupled to downstream signaling components including cGMP. Since during exercise the muscle may utilize insulin-independent mechanisms to increase glucose uptake, the mechanisms involved should provide important knowledge to the understanding and managing of insulin resistance.
Over the past few years many components of the insulin receptor signaling network have been discovered and particular exciting has been the findings that S-nitrosylation-mediated inactivation of Akt/PKB may contribute to the pathogenesis of iNOS- and/or oxidative stress-involved insulin resistance. However, the effect of NO donors on basal and insulin-stimulated glucose uptake in skeletal muscles is controversial. Recent studies demonstrated that low concentrations of NO donors stimulate glucose uptake while higher concentrations are inhibitory. The mechanisms involved have not been fully elucidated and new studies should be designed to define the precise molecular mechanisms.
REFERENCES
- Abu-Soud, H.M. and D.J. Stuehr, 1993. Nitric oxide synthases reveal a role for calmodulin in controlling electron transfer. Proc. Natl. Acad. Sci. USA., 90: 10769-10772.
Direct Link - Agati, J.M., D. Yeagley and P.G. Quinn, 1998. Assessment of the roles of mitogen-activated protein kinase, phosphotidylinositol 3-kinase, protein kinase B and protein kinase C in insulin inhibition of cAMP-induced phosphoenolpyruvate carboxykinase gene transcription. J. Biol. Chem., 273: 18751-18759.
Direct Link - Anderson, K., N. Gaudiot, C. Ribiere, M. Elizalde, Y. Giudicelli and P. Arner, 1999. A nitric oxide-mediated mechanism regulates lipolysis in human adipose tissue in vivo. Br. J. Pharmacol., 126: 1639-1645.
Direct Link - Balon, T.W. and J.L. Nadler, 1997. Evidence that nitric oxide increases glucose uptake in skeletal muscle. J. Applied Physiol., 82: 359-363.
Direct Link - Baron, A.D., G. Brechtel, P. Wallace and S.V. Edelman, 1988. Rates and tissue sites of non-insulin-and insulin-mediated glucose uptake in humans. Am. J. Physiol. Endocrinol. Metab., 255: 769-774.
Direct Link - Baron, A.D., G. Brechtel-Hook, A. Johnson and D. Hardin, 1993. Skeletal muscle blood flow: A possible link between insulin resistance and blood pressure. Hypertension, 21: 129-135.
Direct Link - Baron, A.D., 1994. Hemodynamic actions of insulin. Am. J. Physiol. Endocrinol. Metabol., 267: E187-E202.
Direct Link - Baron A.D., H.O. Steinberg, H. Chaker, R. Leaming, A. Johnson and G. Brechtel, 1995. Insulin-mediated skeletal muscle vasodilation contributes to both insulin sensitivity and responsiveness in lean humans. J. Clin. Invest., 96: 786-792.
Direct Link - Baron, A.D., G. Brechtel-Hook, A. Johnson, J. Cronin, R. Leaming and H.O. Steinberg, 1996. Effect of perfusion rate on the time course of insulin-mediated skeletal muscle glucose uptake. Am. J. Physiol. Endocrinol. Metabol., 271: E1067-E1072.
Direct Link - Baron, A.D. and M.G. Clark, 1997. Role of blood flow in the regulation of muscle glucose uptake. Annu. Rev. Nutr., 17: 487-499.
Direct Link - Bedard, S., B. Marcotte and A. Marette, 1997. Cytokines modulate glucose transport in skeletal muscle by inducing the expression of inducible nitric oxide synthase. Biochem. J., 325: 487-493.
Direct Link - Bedard, U.V., B. Marcotte and A. Marette, 1998. Insulin inhibits inducible nitric oxide synthase in skeletal muscle cells. Diabetologia, 41: 1523-1527.
Direct Link - Begum, N., L. Ragolia, J. Rienzie, M. McCarthy and N. Duddy, 1998. Regulation of mitogen-activated protein kinase phosphatase-1 induction by insulin in vascular smooth muscle cells. J. Biol. Chem., 273: 25164-25170.
Direct Link - Bergandi, L., F. Silvagno, I. Russo, C. Riganti and G. Anfossi et al., 2003. Insulin stimulates glucose transport via nitric oxide/cyclic GMP pathway in human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol., 23: 2215-2221.
Direct Link - Bergemann, C., C. Loken, C. Becker, B. Graf, M. Hamidizadeh and Y. Fisher, 2001. Inhibition of glucose transport by cyclic GMP in cardiomyocytes. Life Sci., 69: 1391-1406.
Direct Link - Boczkowski, J., E. Vicaut, G. Danialou and M. Aubier, 1994. Role of nitric oxide and prostaglandins in the regulation of diaphragmatic arteriolar tone in the rat. J. Applied Physiol., 77: 590-596.
Direct Link - Bradley, S.J., B.A. Kingwell and G.K. McConell, 1999. Nitric oxide synthase inhibition reduces leg glucose uptake but not blood flow during dynamic exercise in humans. Diabetes, 48: 1815-1821.
Direct Link - Butler, R., A.D. Morris and A. Struthers, 1998. Systemic nitric oxide synthase inhibition increases insulin sensitivity in man. Clin. Sci., 94: 175-180.
Direct Link - Carvalho, E., B. Eliasson, C. Wesslau and U. Smith, 2000. Impaired phosphorylation and insulin-stimulated translocation to the plasma membrane of protein kinase B/Akt in adipocytes from Type II diabetic subjects. Diabetologia (Mex), 43: 1107-1115.
Direct Link - Carvalho-Filho, M.A., M. Ueno, S.M., Hirabara, A.B., Seabra and J.B. Carvalheira et al., 2005. S-Nitrosation of the insulin receptor, insulin receptor substrate 1 and protein kinase B/Akt-A novel mechanism of insulin resistance. Diabetes, 54: 959-967.
Direct Link - Ceriello, A., L. Quagliaro, M. D-Amino, C. Di Filippo and R. Marfella et al., 2002. Acute hyperglycaemia induces nitrotyrosine formation and apoptosis in perfused heart from rat. Diabetes, 51: 1076-1082.
Direct Link - Cheatham, B., C.J. Vlahos, L. Cheatham, L. Wang, J. Blenis and C.R. Kahn, 1994. Phosphatidylinositol 3-kinase activation is required for insulin stimulation of pp70 S6 kinase, DNA synthesis and glucose transporter translocation. Mol. Cell Biol., 14: 4902-4911.
Direct Link - Chen, Y.L. and E.J. Messina, 1996. Dilation of isolated skeletal muscle arterioles by insulin is endothelium dependent and nitric oxide mediated. Am. J. Physiol., 270: H2120-H2124.
Direct Link - Clarke, J.F., P.W. Young, K. Yonezawa, M. Kasuga and G.D. Holman, 1994. Inhibition of the translocation of GLUT1 and GLUT4 in 3T3-L1 cells by the phosphatidylinositol 3-kinase inhibitor, wortmannin. Biochem. J., 300: 631-635.
Direct Link - Coderre, L., K.V. Kandror, G. Vallega and P.F. Pilch, 1995. Identification and characterization of an exercise-sensitive pool of glucose transporters in skeletal muscle. J. Biol. Chem., 270: 27584-27588.
Direct Link - Colberg, S.R., J.M. Hagberg, S.D. McCole, J.M. Zmuda, P.D. Thompson and D.E. Kelly, 1996. Utilization of glycogen but not plasma glucose is reduced in individuals with NIDDM during mild-intensity exercise. J. Applied Physiol., 81: 2027-2033.
Direct Link - DeFronzo, R.A., E. Ferrannini, Y. Sato, P. Felig and J. Wahren, 1981. Synergistic interaction between exercise and insulin on peripheral glucose uptake. J. Clin. Invest., 68: 1468-1474.
CrossRefDirect Link - Dela, F., J. Larsen and H. Galbo, 1995. Normal effect of insulin to stimulate blood flow in NIDDM. Diabetes, 44: 221-226.
Direct Link - Depre, C., J.L. Vanoverschelde, J.F. Goudemant, I. Mottet and L. Hue, 1995. Protection against ischemic injury by non-vasoactive concentrations of nitric oxide inhibitors in the perfused rabbit heart. Circulation, 92: 1911-1918.
Direct Link - Depre, C., V. Gaussin, S. Ponchaut, Y. Fisher, J.L. Vanoverschelde and L. Hue, 1998. Inhibition of myocardial glucose uptake by cGMP. Am. J. Physiol., 274: H1443-H1449.
Direct Link - Duplain, H., R. Burcelin and C. Sartori Stephane Cook, Marc Egliet al., 2001. Insulin resistance, hyperlipidemia and hypertension in mice lacking endothelial nitric oxide synthase. Circulation, 104: 342-345.
Direct Link - Elizalde, M., M. Ryden, V. van Harmelen, P. Eneroth and H. Gyllenhammer et al., 2000. Expression of nitric oxide synthase in subcutaneous adipose tissue of non obese and obese humans. J. Lipid Res., 41: 1244-1251.
Direct Link - Etgen, G.J., D.A. Fryburg and E.M. Gibbs, 1997. Nitric oxide stimulates skeletal muscle transport through a calcium/contraction-and phosphotidylinositol-3-kinase-independent pathway. Diabetes, 46: 1915-1919.
Direct Link - Eu, J.P., J. Sun, L. Xu, J.S. Stamler and G. Meissner, 2000. The skeletal muscle calcium release channel: Coupled O2 sensor and NO signaling functions. Cell, 102: 499-509.
Direct Link - Fryer, L., E. Hajduch, F. Rencurel, I. Salt, H. Hundal, D. Hardie and D. Carling, 2000. Activation of glucose transport by AMP-activated protein kinase via stimulation of nitric oxide synthase. Diabetes, 49: 1978-1985.
Direct Link - Goodyear, L.J., F. Giorgino, T.W. Balon, G. Condorelli and R.J. Smith, 1995. Effects of contractile activity on tyrosine phosphoproteins and PI3-kinase activity in rat skeletal muscle. Am. J. Physiol., 268: E987-E995.
Direct Link - Goodyear, L.J., P.Y. Chang, D.J. Sherwood, S.D. Dufresne and D.E. Moller, 1996. Effects of exercise and insulin on mitogen-activated protein kinase signaling pathways in rat skeletal muscle. Am. J. Physiol., 271: E403-E408.
Direct Link - Gow, A.J., Q. Chen, D.T., Hess, B.J. Day, H. Ischiropoulous and J.S. Stamler, 2002. Basal and stimulated protein S-Nitrosylation in multiple cell types and tissues. J. Biol. Chem., 277: 9637-9640.
Direct Link - Greene, M.W., H. Sakaue, L. Wang, D.R. Alessi and R.A. Roth, 2003. Modulation of insulin-stimulated degradation of human insulin receptor substrate-1 by serine 312 phosphorylation. J. Biol. Chem., 278: 8199-8211.
Direct Link - Hardie, D.G. and D. Carling, 1997. The AMP-activated protein kinase: Fuel gauge of the mammalian cell? Eur. J. Biochem., 246: 259-273.
Direct Link - Hayashi, T., M.F. Hirshman, E.J. Kurth, W.W. Winder and L.J. Goodyear, 1998. Evidence for AMP-activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes, 47: 1369-1373.
Direct Link - Hespel, P., L. Vergauwen, K. Vandenberghe and E.A. Richter, 1996. Significance of insulin for glucose metabolism in skeletal muscle contractions. Diabetes, 45: S99-S104.
Direct Link - Hess, D.T., A, Matsumoto, R. Nudelman and J.S. Stamler, 2001. S-nitrosylation: Spectrum and specificity. Nat. Cell Biol., 3: E46-E49.
Direct Link - Higaki, Y., M.F. Hirshman, N. Fujii and L. Goodyear, 2001. Nitric oxide increases glucose uptake through a mechanism that is distinct from the insulin and contraction pathways in rat skeletal muscle. Diabetes, 50: 241-247.
Direct Link - Ignarro, L.J., C. Napoli and J. Loscalzo, 2002. Nitric oxide donors and cardiovascular agents modulating the bioactivity of nitric oxide. Circ. Res., 90: 21-28.
CrossRefDirect Link - Jensen, J., M.N. Sharikabad, K.M. Ostbye, O. Melien and O. Brors, 2003. Evidence that nitroprusside stimulates glucose uptake in cardiomyocytes via mitogen-activated protein kinase. Arch. Physiol. Biochem., 111: 239-245.
Direct Link - Jiang, Z.Y., Q.L. Zhou, K.A. Coleman, M. Chouinard, Q. Boese and M.P. Czech, 2003. Insulin signaling through Akt/protein kinase B analyzed by small interfering RNA-mediated gene silencing. Proc. Natl. Acad. Sci. USA., 100: 7569-7574.
Direct Link - Jun, T. and A. Wennmalm, 1994. NO-dependent and-independent elevation of plasma levels of insulin and glucose in rats by L-arginine. Br. J. Pharmacol., 113: 345-348.
Direct Link - Khan, B.B., 1992. Facilitative glucose transporters: Regulatory mechanisms and dysregulation in diabetes. J. Clin. Invest., 89: 1367-1374.
Direct Link - Kahn, A.M., R.A. Lichtenberg, J.C. Allen, C.L. Seidel and T. Song, 1995. Insulin-stimulated glucose transport inhibits Ca2+ influx and contraction in vascular smooth muscle. Circulation, 92: 1597-1603.
Direct Link - Kapur, S., S. Bedard, B. Marcotte, C.H. Cote and A. Marette, 1997. Expression of nitric oxide synthase in skeletal muscle: a novel role of nitric oxide as a modulator of insulin action. Diabetes, 46: 1691-1700.
Direct Link - Kemp. B.E., K.I. Mitchelhill, D. Stapleton, B.J. Mitchell, Z.P. Chen and L.A. Walters, 1999. Dealing with energy demand: The AMP-activated protein kinase. Trends Biochem. Sci., 24: 22-25.
Direct Link - Kido, Y., J. Nakae and D. Accili, 2001. The insulin receptor and its cellular targets. J. Clin. Endocrinol. Metabol, 86: 972-979.
Direct Link - King, P.A., E.D. Horton, M.F. Hirshman and E.S. Horton, 1992. Insulin resistance in obese Zucker rat (fa/fa) skeletal muscle is associated with a failure of glucose transporter translocation. J. Clin. Invest., 90: 1568-1575.
Direct Link - King, P.A., J.J. Betts, E.D. Horton and E.S. Horton, 1993. Exercise, unlike insulin, promotes glucose transporter translocation in obese Zucker rat muscle. Am. J. Physiol., 265: R447-R452.
Direct Link - Kingwell, B.A., 2000. Nitric oxide-mediated metabolic regulation during exercise: Effects of training in health and cardiovascular disease. FASEB. J., 14: 1685-1696.
Direct Link - Kingwell, B.A., M. Formosa, M. Muhlmann and S.J. Bradley, 2002. Nitric oxide inhibition reduces glucose uptake during exercise in individuals with type 2 diabetes more than in control subjects. Diabetes, 51: 2572-2580.
Direct Link - Kobzik, L., M.B. Reid, D.S. Bredt and J.S. Stamler, 1994. Nitric oxide in skeletal muscle. Nature, 372: 546-548.
Direct Link - Kobzik, L., B. Stringer, J.L. Balligand, M.B. Reid and J.S. Stamler, 1995. Endothelial type nitric oxide synthase in skeletal muscle fibers: Mitochondrial relationships. Biochem. Biophys. Res. Commun., 211: 375-381.
Direct Link - Kurth-Kraczek, E.J., M.F. Hirshman, L.J. Goodyear and W.W. Winder, 1999. AMP-activated protein kinase activation causes GLUT4 translocation in skeletal muscle. Diabetes, 48: 1667-1671.
Direct Link - Kusner, L.L. and H.J. Kaminski, 1996. Nitric oxide synthase is concentrated at the skeletal muscle endplate. Brain Res., 730: 238-242.
Direct Link - Lander, H.M., A.T. Jacovina, R.J. Davis and J.M. Tauras, 1996. Differential activation of mitogen-activated protein kinase by nitric-related species. J. Biol. Chem., 271: 19705-19709.
Direct Link - Lei, B., K. Matsuo, V. Labinskyy, N. Sharma and M.P. Chandler et al., 2005. Exogenous nitric oxide reduces glucose transporters translocation and lactate production in ischemic myocardium in vivo. Proc. Natl. Acad. Sci. USA., 102: 6966-6971.
Direct Link - Li, J., X. Hu, P. Selvakumar, R.R. Russell, S.W. Cushman, G.D. Holman and L.H. Young, 2004. Role of the nitric oxide pathway in AMP-mediated glucose uptake and GLUT-4 translocation in heart muscle. Am. J. Physiol. Endocrinol. Metabol., 287: E834-E841.
Direct Link - Lund, S., G.D. Holman, O. Schmitz and O. Pedersen, 1995. Contraction stimulates translocation of glucose transporter GLUT4 in skeletal muscle through a mechanism distinct from insulin. Proc. Natl. Acad. Sci., 92: 5817-5821.
Direct Link - Marsin, A.S., L. Bertrand, M.H. Rider, J. Deprez and C. Beauloye et al., 2000. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr. Biol., 10: 1247-1255.
Direct Link - Martin, I.K., A. Katz and J. Wahren, 1995. Splanchnic and muscle metabolism during exercise in NIDDM patients. Am. J. Physiol., 269: E583-E590.
Direct Link - McCoy, M.J., J. Proietto and M. Hargreaves, 1996. Skeletal muscle GLUT-4 and post-exercise muscle glycogen storage in humans. J. Applied Physiol., 80: 411-415.
Direct Link - McDonald, L.J. and F. Murad, 1996. Nitric oxide and cyclic GMP signaling. Proc. Soc. Exp. Biol. Med., 211: 1-6.
Direct Link - McGowder, D., D. Ragoobirsingh and T. Dasgupta, 1999. The hyperglycemic effect of S-nitrosoglutathione in the dog. Nitric Oxide, 3: 481-491.
CrossRefDirect Link - McGrowder, D., D. Ragoobirsingh and T. Dasgupta, 2001. Effects of S-Nitroso-N-acetylpenicillamine administration on glucose tolerance and plasma levels of insulin and glucagon in the dog. J. Nitric Oxide-Biol. Chem., 5: 402-412.
Direct Link - McGrowder, D., D. Ragoobirsingh and P. Brown, 2006. Acute effects of exogenous nitric oxide on glucose uptake in skeletal muscle of normoglycaemic and diabetic rats. Med. Sci. Monitor, 12: BR 28-35.
Direct Link - Meneilly, G.S., B. Battistini and J.S. Floras, 2000. Lack of the effect of sodium nitroprusside on insulin-mediated blood flow and glucose disposal in the elderly. Metabolism, 49: 373-378.
Direct Link - Meneilly, G.S., B. Battistini and J.S. Floras, 2001. Contrasting effects of L-arginine on insulin-mediated blood flow and glucose disposal in the elderly. Metabolism, 50: 194-199.
Direct Link - Merrill, G.F., E.J. Kurth, D.G. Hardie and W.W. Winder, 1997. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation and glucose uptake in rat muscle. Am. J. Physiol., 273: E1107-E1112.
Direct Link - Minuk, H.L., M. Vranic, E.B. Marliss, A.K. Hanna, A.M. Albisser and B. Zinman, 1981. Glucoregulatory and metabolic response to exercise in obese noninsulin-dependent diabetes. Am. J. Physiol., 240: E458-E464.
Direct Link - Moncada, S., R.M.J., Palmer and E.A. Higgs, 1991. Nitric oxide physiology, pathology and pharmacology. Pharmacol. Rev., 143: 109-142.
Direct Link - Morrow, V.A., F. Foufelle, J.M.C. Connell, J.R. Petrie, G.W. Gould and I.P. Salt, 2003. Direct activation of AMP-activated protein kinase stimulates nitric oxide synthesis in human aortic endothelial cells. J. Biol. Chem., 278: 31629-31639.
Direct Link - Natali, A., D. Santoro, C. Palombo, M. Cerri, S. Ghione and E. Ferrannini, 1991. Impaired insulin action on skeletal muscle metabolism in essential hypertension. Hypertension, 17: 170-178.
Direct Link - Natali A., A.Q. Galvan, N. Pecori, G. Sanna, E. Toschi and E. Ferrannini, 1998. Vasodilation with sodium nitroprusside does not improve insulin action in essential hypertension. Hypertension, 31: 632-636.
Direct Link - Oshida, Y., Y. Tachi, Y. Morishita, K. Kitakoshi and N. Fuku et al., 2000. Nitric oxide decreases insulin resistance induced by high-fructose feeding. Horm. Metabol. Res., 32: 339-342.
Direct Link - Perreault, M. and A. Marette, 2001. Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat. Med., 7: 1138-1143.
CrossRefDirect Link - Pfeifer, A., P. Ruth, W. Dostmann, M. Sausbier, P, Klatt and F. Hofmann, 1999. Structure and function of cGMP-dependent protein kinases. Rev. Physiol. Biochem. Pharmacol., 135: 105-149.
Direct Link - Piatti, P.M., L.D. Monti, G. Valsecchi, F. Magni and E. Setola et al., 2001. Long-term oral L-arginine administration improves peripheral and hepatic insulin sensitivity in type 2 diabetic patients. Diabetes Care, 24: 875-880.
Direct Link - Pilon, G., P. Penfornis and A. Marette, 2000. Nitric oxide production by adipocytes: A role in the pathogenesis of insulin resistance? Horm. Metabol., 32: 480-484.
Direct Link - Pilon, G., P. Dallaire and A. Marette, 2004. Inhibition of inducible nitric oxide synthase by activation of AMP-activated protein kinase: A new mechanism of action of insulin-sensitizing drugs. J. Biol. Chem., 279: 20767-20774.
CrossRefDirect Link - Pitre, M.A., A. Nadeau and H. Bachelard, 1996. Insulin sensitivity and hemodynamic responses to insulin in Wistar-Kyoto and spontaneously hypertensive rats. Am. J. Physiol., 271: E658-E668.
Direct Link - Ploug, T., H. Galbo and E.A. Richter, 1984. Increased muscle glucose uptake during contractions: No need for insulin. Am. J. Physiol., 247: E726-E731.
Direct Link - Potenza, M.A., F.L. Marasciulo, D.M. Chieppa, G.S. Brigiani, G. Formoso, M. J. Quon and M. Montagnani, 2005. Insulin resistance in spontaneously hypertensive rats is associated with endothelial dysfunction characterized by imbalance between NO and ET-1 production. Am. J. Physiol. (Heart Circ.) Physiol., 289: H813-H822.
Direct Link - Ribiere, C., A.M. Jaubert, N. Gaudiot, D. Sabourault and M.L. Marcus et al., 1996. White adipose tissue nitric oxide synthase: A potential source for NO production. Biochem. Biophys. Res. Commun., 222: 706-712.
Direct Link - Roberts, C.K., R.J. Barnard, S.H. Scheck and T.W. Balon, 1997. Exercise-stimulated glucose transport in skeletal muscle is nitric oxide dependent. Am. J. Physiol., 273: E220-E225.
Direct Link - Roy, D., M. Perreault and A. Marette, 1998. Insulin stimulation of glucose uptake in skeletal and adipose tissues in vivo is NO dependent. Am. J. Physiol., 274: E692-E699.
Direct Link - Russell, R.R., J. Li, D.L. Coven, M. Pypaert and C. Zechner et al., 2004. AMP-activated protein kinase mediates ischaemic glucose uptake and prevents post-ischaemic cardiac dysfunction, apoptosis and injury. J. Clin. Invest., 114: 495-503.
Direct Link - Salt, I.P., J.M. Connell and G.W. Gould, 2000. 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR) inhibits insulin-stimulated glucose transport in 3T3-L1 adipocytes. Diabetes, 49: 1649-1656.
Direct Link - Saltiel, A.R. and C.R. Khan, 2001. Insulin signaling and the regulation of glucose and lipid metabolism. Nature, 414: 799-806.
Direct Link - Sarabi, M., L. Lind, J. Millgard, A. Hanni, A. Hagg, C. Berne and H. Lithell, 1999. Local vasodilation with metacholine, but not with nitroprusside, increases forearm glucose uptake. Physiol. Res., 48: 291-295.
Direct Link - Scherrer, U., D. Randin, P. Vollenweider, L. Vollenweider and P. Nicod, 1994. Nitric oxide release accounts for insulin's vascular effects in humans. J. Clin. Invest., 94: 2511-2515.
CrossRefDirect Link - Scherrer, U. and C. Sartori, 2000. Defective nitric oxide synthesis: A link between metabolic insulin resistance, sympathetic over-activity and cardiovascular morbidity. Eur. J. Endocrinol., 142: 315-323.
Direct Link - Shankar, R., J.S. Zhu, B. Ladd, D. Henry, H.Q. Shen and A.D. Baron, 1998. Central nervous system nitric oxide synthase activity regulates insulin secretion and action. J. Clin. Invest., 102: 1403-1412.
Direct Link - Shankar, R.R., Y. Wu, H.Q. Shen, J.S. Zhu and A.D. Baron, 2000. Mice with gene disruption of both endothelial and neuronal nitric oxide synthase exhibit insulin resistance. Diabetes, 49: 684-687.
Direct Link - Shao, J., H. Yamashita, L. Qiao and J.E. Friedman, 2000. Decreased Akt kinase activity and insulin resistance in C57BL/KsJ-Leprdb/db mice. J. Endocrinol., 167: 107-115.
Direct Link - Sharma, K., T.M. Danoff, A. DePiero and F.N. Ziyadeh, 1995. Enhanced expression of inducible nitric oxide synthase in murine macrophages and glomerular mesangial cells by elevated glucose levels: Possible mediation via protein kinase C. Biochem. Biophys. Res. Commun., 207: 80-88.
Direct Link - Shearer, J., P.T. Fueger, B. Vorndick, D.P. Bracy, J.N. Rottman, J.A. Clanton and D.H. Wasserman, 2004. AMP kinase-induced skeletal muscle glucose but not long-chain fatty acid uptake is dependent on nitric oxide. Diabetes, 53: 1429-1435.
Direct Link - Stamler, J.S., S. Lamas and F.C. Fang, 2001. Nitrosylation. The prototypic redox-based signaling mechanism. Cell, 106: 675-683.
Direct Link - Standley, P.R., K.A. Rose and J.R. Sowers, 1995. Increased basal arterial smooth muscle glucose transport in the Zucker rat. Am. J. Hypertens., 8: 45-52.
Direct Link - Steinberg, H.O., G. Brechtel, A. Johnson, N. Fineberg and A.D. Baron, 1994. Insulin-mediated skeletal muscle vasodilation is nitric oxide dependent. A novel action of insulin to increase nitric oxide release. J. Clin. Invest., 94: 1172-1179.
Direct Link - Sugita, H., M. Fujimoto, T. Yasukawa, N. Shimizu and M. Sugita et al., 2005. Inducible nitric oxide synthase and NO donor induce insulin receptor substrate-1 degradation in skeletal muscle cells. J. Biol. Chem., 280: 14203-14211.
Direct Link - Tada, H., C.I. Thompson, F.A. Recchia, K.E. Loke and M. Ochoa et al., 2000. Myocardial glucose uptake is regulated by nitric oxide via endothelial nitric oxide synthase in Langendorff mouse heart. Cir. Res., 86: 270-274.
Direct Link - Tanaka, T., K. Nakatani, K. Morioka, H. Urakawa and N. Maruyama et al., 2003. Nitric oxide stimulates glucose transport through insulin-dependent GLUT4 translocation in 3T3-L1 adipocytes. Eur. J. Endocrinol., 149: 61-67.
Direct Link - Torres, S.H., J.B. De Sanctis, L.M. de Briceno, N. Hernandez and H.J. Finol, 2004. Inflammation and nitric oxide production in skeletal muscle of type 2 diabetic patients. J. Endocrinol., 181: 419-427.
CrossRefDirect Link - Trovati, M. and G. Anfossi, 2002. Influence of insulin and of insulin resistance on platelet and vascular smooth muscle cell function. J. Diab. Complic., 16: 35-40.
Direct Link - Trovati, M., P. Massucco, L. Mattiello, F. Cavalot, E. Mullaroni, A. Hahn and G. Anfossi, 1995. Insulin increases cyclic nucleotide content in human vascular smooth muscle cells: A mechanism potentially involved in insulin-induced modulation of vascular tone. Diabetologia, 38: 936-941.
Direct Link - Vallance, P., J. Collier and S. Moncada, 1989. Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet, 334: 997-1000.
CrossRefDirect Link - White, M.F. and C.R. Kahn, 1994. The insulin signaling system. J. Biol. Chem., 269: 1-4.
PubMedDirect Link - Winder, W.W. and D.G. Hardie, 1999. AMP-activated protein kinase, a metabolic master switch: Possible roles in type 2 diabetes. Am. J. Physiol., 277: E1-E10.
CrossRefDirect Link - Yasukawa, T., E. Tokunaga, H. Ota, H., Sugita, J.A. Martyn and M. Kaneki, 2005. S-nitrosylation-dependent inactivation of Akt/Protein kinase B in insulin resistance. J. Biol. Chem., 280: 7511-7518.
Direct Link - Young, L.H., R.F. Yin, R.R. Russell, X. Hu, M. Caplan, J. Ren, G.I. Shulman and A.J. Sinusas, 1997. Low flow ischemia leads to translocation of canine heart GLUT-4 and GLUT-1 glucose transporters to the sarcolemma in vivo. Circulation, 95: 415-422.
Direct Link - Young, M.E., G.K. Radda and B. Leighton, 1997. Nitric oxide stimulates glucose transport and metabolism in rat skeletal muscle in vitro. Biochem. J., 322: 223-228.
Direct Link - Young, M.E. and B. Leighton, 1998. Evidence for altered sensitivity of the nitric oxide/cGMP signaling cascade in insulin-resistant skeletal muscle. Biochem. J., 329: 73-79.
Direct Link - Zierath, J.R., L. He, A. Guma, E. W. Odegoard, A. Klip and H. Wallberg-Henriksson, 1996. Insulin action on glucose transport and plasma membrane GLUT4 content in skeletal muscle from patients with NIDDM. Diabetologia, 39: 1180-1189.
Direct Link