
R. Sharma
Department of Microbiology, University of Delhi, South Campus, New Delhi 110021, India
R. Gupta
Department of Microbiology, University of Delhi, South Campus, New Delhi 110021, India
Research Journal of Microbiology
Year: 2010 | Volume: 5 | Issue: 10 | Page No.: 954-965







ABSTRACT
Fifty proteolytic bacterial cultures were screened for their potential to degrade chicken feather. Eight strains which degraded chicken feather within 24 h were further evaluated for degradation of surrogate yeast prion protein, Sup35 NM. Prion degradation was studied by congo red binding assay and a strain of Pseudomonas aeruginosa KS-1 was selected for further evaluation as it exhibited maximal shift in absorbance of congo red from 0.268 to 0.926. Keratinase from this strain was purified using Q-sepharose chromatography with 60% recovery and a purification fold of 2.24. It was identified as a 33 kDa monomeric protein having a pH and temperature optima of 7 and 50°C, respectively. It was stable over a broad range of pH ranging from pH 2-11 and was highly thermostable with a t1/2 of >2 h at 80°C. It was a serine, thiol activated protease with two fold increase in activity in presence of β-mercaptoethanol. It also hydrolyzed a range of complex protein substrates including casein, feather, fibrin. Amidolytic activity on synthetic substrates revealed that it efficiently cleaved phenylalanine and arginine. It hydrolyzed insulin B-chain at cystein, glycine, histidine, phenylalanine and leucine as revealed by ESI-MS analysis.
PDF Abstract XML References Citation
How to cite this article
R. Sharma and R. Gupta, 2010. Thermostable, Thiol Activated Keratinase from Pseudomonas aeruginosa KS-1 for Prospective Applications in Prion Decontamination. Research Journal of Microbiology, 5: 954-965.
URL: https://scialert.net/abstract/?doi=jm.2010.954.965
URL: https://scialert.net/abstract/?doi=jm.2010.954.965
INTRODUCTION
Keratins are hard-to-degrade, structural proteins that comprise 90% of the dry weight of feathers (Tatineni et al., 2008). They are resistant to degradation by a broad range of proteases like trypsin, pepsin and papain because of their tight secondary structure and presence of disulfide linkages (Pillai and Archana, 2008). However, despite of their rigidity they are efficiently degraded by a compendium of bacteria, actinomycetes and fungi that elaborate keratinase (Gupta and Ramnani, 2006, Saber et al., 2010). Keratinases from gram positive bacteria have been studied in great detail especially from Bacillus licheniformis, however, scanty information is available about keratinases from gram negative bacteria (Riffel et al., 2003). By the virtue of their ability to attack keratins, they not only find application in traditional industries like detergent, leather and feed but are rapidly gaining importance in newer fields like prion degradation, feather meal production, fabrication of bleach free paper, manufacture of glues, films etc. (Brandelli et al., 2009; Kumar et al., 2008). The current focus in keratinase research is shifting towards the challenging field of prion degradation. Prion proteins pose serious problems as they cause Transmissible Spongiform Encephalopathies (TSE) including mad cow disease, sheep scrapie, deer chronic wasting disease, Bovine Spongiform Encephalopathy (BSE) in cattle and Creutzfeldt-Jakob disease in humans. The highly aggregated beta sheet structure of prions is similar to that of feather and thus it is hypothesized that keratinases which are capable of degrading feather would also be instrumental in degradation of prion proteins as well as other beta amyloids (Tsiroulnikov et al., 2004). Therefore, there is a growing trend for screening of keratinolytic microorganisms for evaluating their prion degrading potential. In this respect, keratinases from Bacillus licheniformis, Streptomyces sp. and Nocardiopsis sp. TOA-1 have been reported to degrade prion (Langeveld et al., 2003; Hui et al., 2004; Mitsuiki et al., 2006). In the present study, we attempted to evaluate fifty keratinolytic bacterial cultures for their potential to degrade prion. In preliminary screening, fast degraders which could degrade chicken feather within 24 h were shortlisted. Among them, Pseudomonas aeruginosa KS-1 was finally selected as it showed maximal degradation of yeast surrogate prion protein, Sup35 NM, as studied by congo red binding assay. Purification and detailed biochemical characterization of its keratinase has also been reported.
MATERIALS AND METHODS
The present study was carried out at Department of Microbiology, University of Delhi from October 2008 to January 2010.
Evaluation of Feather Degrading Potential
Fifty feather degrading bacterial cultures were procured from the laboratory culture collection. The isolates were revived from glycerol stocks (50% v/v) in nutrient broth at 37°C, 200 rpm. They were screened for their ability to completely degrade chicken feather in feather peptone medium at 37°C, 200 rpm for upto five days (Ramnani and Gupta, 2004). Eight cultures which completely degraded feather within 24 h were selected for further evaluation for prion degradation. These cultures were identified by Biomeriux/ 16S gene sequencing.
Keratinase Production
For keratinase production, a modified feather –peptone medium was used (Ramnani and Gupta, 2004). Two percent of 24 h old cultures grown in nutrient broth were used as inoculum. The flasks were incubated at 37°C under shaking (250 rpm) in a New Brunswick Scientific shaker (Edison, New Jersey). After 24 h of incubation, the cells were harvested by centrifugation at 8000 rpm (Sigma Centrifuge, Germany) for 10 min. The supernatant was microfiltered using 0.2 μ filters (mdi, India) and concentrated by lyophilization. This preparation was used as keratinase sample for prion degradation.
Keratinase Assay and Protein Determination
Keratinase activity was measured as described by Dozie et al. (1994) with some modifications. The assay mixture containing 1 mL of appropriately diluted enzyme, 4 mL glycine-NaOH buffer (0.05 mM, pH 10) and 20 mg of feather was incubated at 60°C for 60 min. The reaction was terminated by adding 4 mL of 5% (w/v) trichloroacetic acid and the tubes were incubated at room temperature (25°C) for 1 h. Feather and insoluble residues were removed by filtration through glass wool and the filtrate was centrifuged at 5000 g for 5 min. Appropriate enzyme and substrate controls were also set up. Proteolytic products in the supernatant were determined by absorbance at 280 nm against controls. An increase in absorbance of 0.01 was considered as 1 U enzyme activity. The protein concentration was determined by the method of Bradford (1976) taking Bovine Serum Albumin (BSA) as standard. The protein concentration was determined spectrophotometrically at 595 nm.
Caseinolytic Assay
The proteolytic activity was assayed at 60°C in glycine-NaOH buffer (0.05 mol L-1, pH 10) as described previously (Beg and Gupta, 2003). One milliliter of appropriately diluted enzyme was incubated with 1 mL of casein solution prepared in glycine-NaOH, buffer pH 10 for 10 min at 60°C. The reaction was stopped by the addition of 4 mL of 5% (w/v) trichloroacetic acid. The contents were centrifuged after 1 h at 8000 rpm for 10 min. Folin Ciocalteau's reagent (0.5 mL) was added to 1 mL of the supenatant and the optical density of the samples was taken at 660 nm against appropriate substrate and enzyme blanks. One unit of protease was equivalent to the amount of enzyme required to release 1 μg of tyrosine mL-1 min-1 under standard assay conditions.
Overexpression and Purification of Yeast Surrogate Prion Protein
The plasmid of yeast surrogate prion (Sup35 NM) protein was purchased from North Carolina University, USA. It was transformed into E. coli BL21 CODON PLUS cells. The positive clones were stored in 50% glycerol at -80°C. It was revived in LB containing 100 mg mL-1 ampicillin at 37°C, 200 r pm for overnight. 1% of this culture was grown in LB-Amp till an OD600 of 0.5 was reached. 1 mM of IPTG was then added to the culture to induce expression of prion protein at 37°C, 200 rpm for 3 h. After induction, the cells were harvested and resuspended in lysis buffer (10 mM Tris-HCl, pH 7.2, 1 mM DTT, 1 mM PMSF, 8 M urea) and sonicated by 9 sec pulse on and 9 sec pulse off for 2 min. Sonication was followed by centrifugation at 5000 rpm for 30 min. The level of expression was checked on 15% SDS-PAGE. Sup 35 NM was purified a s described by Chen et al. (2005) using Q-Sepharose and hydroxyapetite chromatography. The purified protein was concentrated by adding 50% chilled methanol followed by centrifugation at 12,000 rpm for 15 min. The precipitate was stored at -80°C for further use.
Enzymatic Degradation of Sup35 NM
First the congo red assay was standardized by studying the aggregation and deaggregation of Sup 35 NM. Twenty five milligram of methanol precipitated Sup35 NM was resuspended in Congo Red Binding Buffer (CRBB) (5 mM potassium phosphate [pH 7.4], 150 mM NaCl). To study its aggregation and deaggregation, 200 μL of the suspended Sup35 NM was incubated at temperatures ranging from 10 to 90°C for 1h. Congo red binding assay was carried out as described previously (Klunk et al., 1989). Sup 35 NM was mixed with 10 μM of congo red (Sigma Aldrich, USA) prepared in CRBB buffer and its absorbance was monitored between 190 and 600 nm (UV-1800 Shimadzu Spectrophotometer, UV-Probe Ver. 2.3 software).
Five hundred units of keratinase of each of the selected isolate was added to 25 mg of the Sup35 NM resuspended in CRBB and it was incubated at 37°C for 16 h. After incubation, 10 μM congo red was added and absorbance was monitored as described above.
Purification of Keratinase
The cell free culture broth was first lyophilized. One gram of the lyophilized enzyme was dissolved in 5 mL of 50 mM Tris-HCl buffer (pH 8) and loaded on Q-Sepharose column (Sigma Aldrich, USA) equilibrated with 50 mM Tris-HCl buffer (pH 8). The column was washed with the same buffer and 15 mL fraction was collected at a flow rate of 2 mL min-1. Bound protein was eluted in a linear salt gradient (0.1-1M NaCl). Twenty fractions of 2 mL each were collected for each salt concentration. Protein elution was monitored by measuring absorbance at 280 nm and caseinolytic activity was determined as described earlier. Purity of the protein was determined by SDS-PAGE analysis.
Zymogram Analysis
The zymogram of the purified keratinase was carried out with slight modification of the protocol followed by Najafi et al. (2005). The keratinase samples were mixed with the electrophoresis sample buffer without heat denaturation prior to electrophoresis. Native-PAGE was carried out using 10% polyacrylamide gel. After electrophoresis, the gel was washed with glycine-NaOH buffer (50 mM, pH 10.0) for 10 min on a rocker. The gel slab was then overlaid onto a 1% w/v casein agar plate prepared in glycine-NaOH buffer (50 mM, pH 10.0) and incubated at 60°C for 18 h. After incubation, the plate was flooded with 5% trichloroacetic acid.
Biochemical Characterization of Keratinase Effect of pH and Temperature on Keratinase
The effect of pH on the keratinolytic activity was studied by carrying out the keratinase assay in the pH range of 3.0-12.0 using buffers (0.05 M) of varying pH (citrate phosphate buffer (pH 3.0-6.0), sodium phosphate buffer (pH 7.0), tris-HCl buffer (pH 8.0-9.0), glycine-NaOH buffer (pH 10.0), phosphate hydroxide buffer (pH 11.0), hydroxide-chloride buffer (pH 12.0). Similarly, the effect of temperature on keratinase activity was determined by incubating the reaction mixture at different temperatures ranging from 40-70°C at pH 7. Activity was expressed as percentage relative activity with respect to maximum activity, which was considered as 100%.
Effect of pH and Temperature on the Stability of Keratinase
The pH stability was determined by incubating the enzyme in buffers of varying pH (3.0-12.0) for 1 h at room temperature (25±1°C) and thereafter the residual activity was determined at the optimum pH and temperature. The temperature stability was determined by incubating the enzyme samples at various temperatures ranging from 50-80°C for different time intervals upto 2 h (30, 60, 90 and 120 min). The residual activity was determined at pH 7 and 50°C.
Effect of Inhibitors on Keratinase
The purified keratinase was pre-incubated at room temperature (25±1°C) for 15 min with various inhibitors viz., Phenyl methyl sulfonyl fluoride (PMSF), ethylenediamine tetraacetate (EDTA), bromoacetic acid, iodoacetic acid, β-mercaptoethanol, dithiothreitol (DTT), Trypsin inhibitor, 1,10-Phenanthroline, 5,5'-Dithio-bis(2-nitrobenzoic acid) (DTNB), Phosphorhamidon, Bestatin (Sigma-Aldrich, USA; ICN chemicals, USA) at a final concentration of 10 mM.
Substrate Specificity of Keratinase Proteolytic Activity on Complex Substrates
The proteolytic activity of the keratinase was studied using both soluble and insoluble substrates. Complex substrates included casein, gelatin, elastin, feather keratin, fibrin, nail keratin (substrates were either locally available from CDH and SRL or purchased from Sigma-Aldrich, USA). Twenty milligram of each of the substrates was added to 1 mL of appropriately diluted enzyme prepared in sodium phosphate buffer (50 mM, pH 7) and it was incubated at 50°C for 1h. The products released were measured by Folin Ciocalteau's reagent as described earlier.
Amidolytic Activity on Synthetic Substrates
Amidolytic activity was examined using synthetic p-nitroanilide substrates viz., N-Suc-Ala-Ala-Pro-Phe-pNA (AAPF), N-Suc-Ala-Ala-Ala-pNA (AAA), N-Bz-Tyr-pNA, N-Bz-Phe-pNA, N-Bz-Arg-pNA, N-Suc-Ala-Ala-Pro-Leu-pNA, N-Suc-Gly-Gly-Phe-pNA (Sigma-Aldrich, USA; ICN chemicals, USA). Activity was also checked on the synthetic substrate of plasmin i.e., D-Val-Leu-Lys-pNA. The 1 mM of each substrate was added to 1 mL of appropriately diluted enzyme prepared in sodium phosphate buffer (50 mM, pH 7). The reaction mix was incubated at optimum temperature for 10 min. The hydrolyzed product was measured at 405 nm using a UV-Vis spectrophotometer (UV 1700 Shimadzu, Japan). The molar extinction coefficient for pNA was taken to be 9900 M-1cm-1.
Hydrolysis of Insulin B Chain and Mass Spectrometry
The substrate specificity of the keratinase was also determined on the basis of hydrolysis of insulin B-chain (Sigma, cysteine residues oxidized). Hundred microliter of purified enzyme was incubated with 100 μL of insulin B-chain (1 mg mL-1 in sodium phosphate buffer, 10 mM, pH 9). The mixture was incubated at 50°C for 16 h, after which 40 μL of 0.1% (v/v) TFA was added to inactivate the enzyme. Identification of the cleavage products was performed by liquid chromatography-electronspray mass spectrometry (LC-ESI/MS, GenPro Biotech, India). The m/z values ranged between 650 and 2100. Hydrolysis sites were determined using the FindPept, a part of the Expasy software package.
RESULTS AND DISCUSSION
Selection of Prospective Prion Degrading Bacterium
Among fifty feather degrading bacterial cultures, eight strains degraded chicken feather completely within 24 h. These were identified as different species of Bacillus and a strain of Pseudomonas aeruginosa KS-1. Bacillus species are well reported to degrade feather efficiently with varying potential. However, only a few gram negative keratinase producers are known namely Vibrio sp., Chryseobacterium sp., Burkholderia sp. and Pseudomonas aeruginosa (Riffel and Brandelli, 2006; Lin et al., 2009). Further, feather degrading potential of a keratinase indicates their prospective role in prion degradation as the β-pleated structure of both the proteins is highly analogous (Hui et al., 2004; Yoshioka et al., 2007). Keratinases from the selected strains were evaluated for the degradation of yeast surrogate prion protein Sup35 NM by congo red assay. Congo red binds to the amyloid structure of the aggregated prion (Chen et al., 2005). When there is degradation of prion protein, there is release of free congo red resulting in subsequent increase in absorbance. Among the eight strains, keratinase from Bacillus sp. KS-5 (Fig. 1b), Bacillus subtilis KS-9 (Fig. 1c), Bacillus pumilus KS-12 (Fig. 1d), Bacillus licheniformis KS-13 (Fig. 1e), Bacillus megaterium KS-15 (Fig. 1f) and Bacillus sp. KS-90 (Fig. 1g) resulted in marginal changes in congo red absorbance upon incubation with Sup35 NM at 37°C for 16 h indicating that they did not degrade prion protein. However, rest of the two strains namely Pseudomonas aeruginosa KS-1 (Fig. 1a) and Bacillus pumilus KS-98 (Fig. 1h) resulted in significant shift in absorbance of congo red from 0.268 to 0.926 and from 0.268 to 0.479, respectively indicating that both the strains were capable of attacking prion protein with maximum degradation in case of Pseudomonas aeruginosa KS-1.
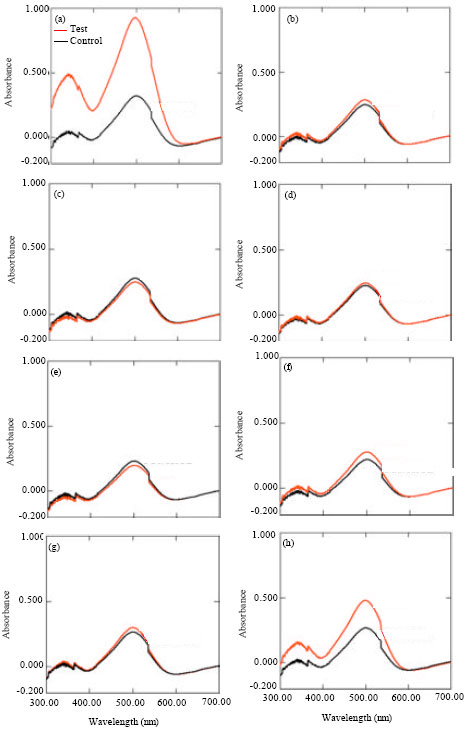 | |
| Fig. 1: | Elution profile of keratinase on Q-sepharose column at pH 8. (a) Pseudomonas aeruginosa KS-1, (b) Bacillu sp. KS-5, (c) Bacillus subtilis KS-9, (d) Bacillus pumilus KS-12, (e) Bacillus licheniformis KS-13, (f) Bacillus megaterium KS-15, (g) Bacillus sp. and (h) Bacillus pumilus KS-98 |
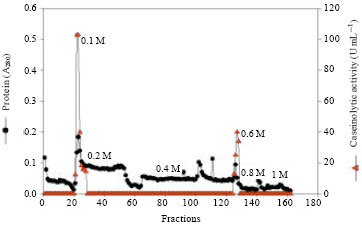 | |
| Fig. 2: | Elution profile of keratinase on Q sepharose column at pH 8 |
 | |
| Fig. 3: | (a-c) SDS-PAGE, Native PAGE and zymogram analysis of purified keratinase |
| Table 1: | Purification scheme of keratinase |
 | |
Hence, detailed biochemical characterization of keratinase from Pseudomonas aeruginosa KS-1 was undertaken in the present study.
Purification of Keratinase
Single step purification of the keratinase was achieved using Q-sepharose column. The present keratinase was eluted in 0.1 M NaCl fraction with a purification fold of 2.24 (Fig. 2, Table 1). The homogeneity of the purified keratinase was revealed by SDS-PAGE, showing a single protein band at 33 kDa (Fig. 3a-c). Usually the molecular weight of keratinases from common keratinolytic bacteria including Bacillus and Streptomyces sp. is around 20-50 kDa (Tatineni et al., 2008). On zymogram analysis the band for keratinase appeared as a translucent zone against a white background (Fig. 3).
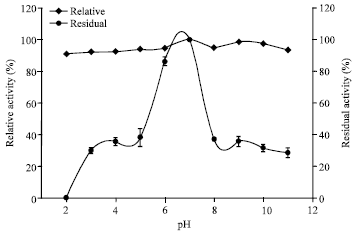 | |
| Fig. 4: | Effect of pH on the activity and stability of keratinase |
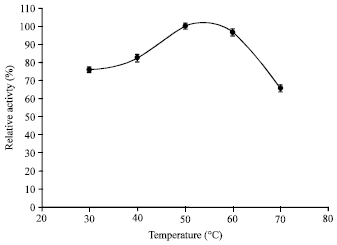 | |
| Fig. 5: | Effect of temperature on activity of keratinase at pH 7 |
Effect of pH on the Activity and Stability of Keratinase
Biochemical characterization of the keratinase revealed it to be a neutral protease with a sharp optima at pH 7 (Fig. 4). It also had high stability over a broad range of pH varying from pH 5 to 9 with 88% residual activity at pH 5 (Fig. 4). This is in contrast to the earlier reported 33 kDa keratinase from Pseudomonas aeruginosa which was an alkaline protease with pH optima of 8 and stability within the range of pH 6-9 (Lin et al., 2009).
Effect of Temperature on the Activity and Stability of Keratinase
The present keratinase was active over a wide temperature range with more than 70% relative activity over a range of 30 to 70°C and maximal activity at 50°C (Fig. 5). Noteworthy, was its thermostability with a t1/2 of more than 2 h at 80°C (Fig. 6). Such high temperature stability is not common in keratinases and it was also not observed in case of earlier reported keratinase from Pseudomonas aeruginosa which had temperature stability within the range of 10-50°C (Lin et al., 2009). This broad range of stability of the present keratinase confers it a potential to be used in various industrial sectors.
| Table 2: | Effect of inhibitors on the activity of keratinase |
 | |
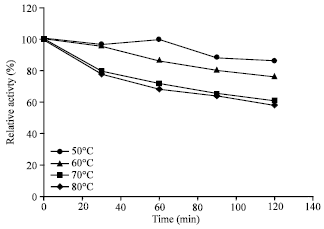 | |
| Fig. 6: | Effect of temperature on stability of keratinase |
Effect of Inhibitors on Keratinase
The effect of various protease inhibitors and chelators on keratinase activity is shown in Table 2. The keratinase was a serine protease as it was inhibited by PMSF. This is in confirmation with most of the reported keratinases where largely they are serine proteases with few exceptions of metalloproteases (Brandelli, 2008). Noteworthy was that it was thiol activated with nearly two fold increase in activity in presence of â- mercaptoethanol. Thiol activation adds onto the potential of a keratinase as feathers are rich in cysteine, their degradation releases thiols which in turn activate the enzyme. It is a common feature which has also been reported for keratinases from other bacteria such as Bacillus subtilis, Stenotrophomonas sp. (Cai et al., 2008; Cao et al., 2009).
Substrate Specificity of Keratinase
The present keratinase exhibited broad substrate specificity as it hydrolyzed a variety of substrates viz., feather keratin, fibrin, meat protein, nail keratin, gelatin with maximal activity on casein (Fig. 7). Its keratinolytic: caseinolytic (K: C) activity was nearly 0.5 conferring the accepted convention for keratinases (Pillai et al., 2008). Amidolytic activity on p-nitroanilides showed preference for phenylalanine and arginine (Fig. 8). It also exhibited 96% relative activity on plasmin substrate i.e., D-Val-Leu-Lys-pNA. High activity on fibrin as well as on synthetic substrate of plasmin suggests that the present keratinase can also be exploited as an efficient fibrinolytic enzyme.
The substrate specificity was also substantiated by hydrolysis of oxidized B-chain of insulin. It showed broad specificities as it hydrolyzed peptide bonds after hydrophobic and negatively charged amino acid resides in the insulin chain (Fig. 9a, b).
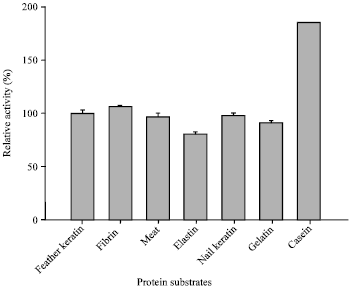 | |
| Fig. 7: | Substrate specificity of keratinase on complex substrates |
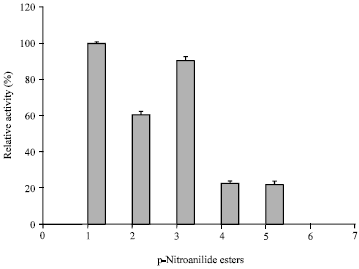 | |
| Fig. 8: | Amidolytic activity of keratinase on synthetic substrate. 1: N-Suc-Ala-Ala-Pro-Phe-pNA, 2: N-Suc-Ala-Ala-pNA, 3: N-benzoyl-DL-Arg-pNA, 4: 1: N-Suc-Ala-Ala-Pro-Leu-pNA, 5: N-Benzoyl-L-tyrosine-pNA |
Residues of Cys are oxidized and this creates two additional negative charges. Besides cleavage at oxidized Cys, the additional hydrolytic sites were identified to be Phe1-Val2, Cys7-Gly8, Gly8-Ser9, Leu11-Val12. It exhibited much broader specificity as compared to subtilisin Carlsberg and subtilisin Novo as it could also cleave the bonds between His10-Leu11 and Phe24-Phe25. Such broad amino acid specificity is in confirmation with the Streptomyces sp. protease which has already been documented for prion degradation justifying Pseudomonas aeruginosa KS-1 keratinase to be a prospective prion degrading enzyme.
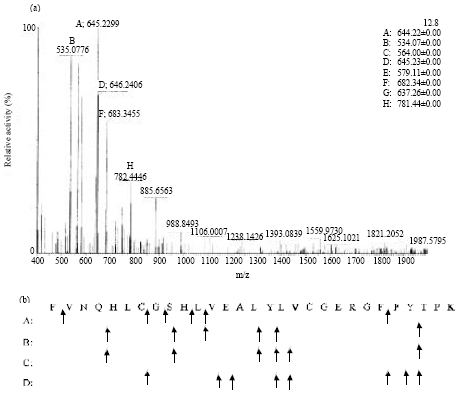 | |
| Fig. 9: | Hydrolysis of insulin B-chains (with oxidized cysteine residues) by keratinase. (a) ESI-LC-MS/MS data (b) Sequence of insulin B-chain. Arrows point at cleavage sites |
ACKNOWLEDGMENT
R. Sharma would like to thank University Grant Commission (UGC) for providing her with Junior Research Fellowship.
REFERENCES
- Beg, Q.K. and R. Gupta, 2003. Purification and characterization of an oxidation-stable, thiol-dependent serine alkaline protease from Bacillus mojavensis. Enzyme Microbial Technol., 32: 294-304.
CrossRef - Brandelli, A., 2008. Bacterial keratinases: Useful enzymes for bioprocessing agroindustrial wastes and beyond. Food Bioprocess Technol., 1: 105-116.
CrossRefDirect Link - Brandelli, A., D.J. Daroit and A. Riffel, 2009. Biochemical features of microbial keratinases and their production and applications. Applied Microbiol. Biotechnol., 85: 1735-1750.
CrossRefPubMedDirect Link - Cai, C.G., J.S. Chen, J.J Qi, Y. Yin and X.D. Zheng, 2008. Purification and characterization of keratinase from a new Bacillus subtilis strain. J. Zheijang Univ. Sci. B, 9: 713-720.
CrossRefDirect Link - Cao, Z.J., Q. Zhang, D.K. Wei, L. Chen, J. Wang, X.Q. Zhang and M.H. Zhou, 2009. Characterization of a novel Strenotrophomonas isolate with high keratinase activity and purification of the enzyme. J. Ind. Microbiol. Biotech., 36: 181-188.
CrossRefDirect Link - Chen, C.Y., K. Rojanatavorn, A.C. Clark and J.C.H. Shih, 2005. Characterization and enzymatic degradation of Sup35NM, a yeast prion-like protein. Protein Sci., 14: 2228-2235.
CrossRefDirect Link - Dozie, I.N.S., C.N. Okeko and N.C. Unaeze, 1994. A thermostable alkaline active, keratinolytic proteinase from Chrytosporium keratinophilum. World J. Microbiol. Biotechnol., 10: 563-567.
CrossRefDirect Link - Gupta, R. and P. Ramnani, 2006. Microbial keratinases and their prospective applications: An overview. Applied Microbiol. Biotechnol., 70: 21-33.
CrossRefDirect Link - Hui, Z., H. Doi, H. Kanouchi, Y. Matsuura, S. Mohri, Y. Nonomura and T. Oka, 2004. Alkaline serine protease produced by Streptomyces sp. degrades PrPSc. Biochem. Biophys. Res. Commun., 321: 45-50.
PubMedDirect Link - Klunk, W.E., J.W. Pettegrew and D.J. Abraham, 1989. Two simple methods for quantifying low affinity dye-substrate binding. J. Histochem. Cytochem., 37: 1293-1297.
PubMedDirect Link - Kumar, D., Savitri, N. Thakur, R. Verma and T.C. Bhalla, 2008. Microbial proteases and application as laundry detergent additive. Res. J. Microbiol., 3: 661-672.
CrossRefDirect Link - Langeveld, J.P.M., J.J. Wang, D.F.M. van de Wiel, G.C. Shih, J. Garssen, A. Bossers and J.C.H. Shih, 2003. Enzymatic degradation of prion protein in brain stem from infected cattle and sheep. J. Infect. Dis., 188: 1782-1789.
Direct Link - Lin, H.H., L.J. Yin and S.T. Jiang, 2009. Functional expression and characterization of keratinase from Pseudomonas aeruginosa in Pichia pastoris. J. Agric. Food Chem., 57: 5321-5325.
CrossRefDirect Link - Mitsuiki, S., Z. Hui, D. Matsumoto, M. Sakai and Y. Moriyama et al., 2006. Degradation of PrPSc by keratinolytic protease from Nocardiopsis sp. TOA-1. Biosci. Biotechnol. Biochem., 70: 1246-1248.
PubMedDirect Link - Najafi, M.F., D. Deobagkar and D. Deobagkar, 2005. Potential application of protease isolated from Pseudomonas aeruginosa PD100. Electronic J. Biotechnol., 8: 197-203.
Direct Link - Pillai, P. and G. Archana, 2008. Hide depilation and feather disintegration studies with keratinolytic serine protease from a novel Bacillus subtilis isolate. Applied Microbiol. Biotechnol., 78: 643-650.
CrossRefPubMedDirect Link - Ramnani, P. and R. Gupta, 2004. Optimization of medium composition for keratinase production on feather by Bacillus licheniformis RG 1 using statistical methods involving response surface methodology. Biotechnol. Applied Biochem., 49: 191-196.
PubMedDirect Link - Riffel, A. and A. Brandelli, 2006. Keratinolytic bacteria isolated from feather waste. Braz. J. Microbiol., 37: 395-399.
CrossRefDirect Link - Riffel, A., F. Lucas, P. Heeb and A. Brandelli, 2003. Characterization of a new keratinolytic bacterium that completely degrades native feather keratin. Arch. Microbiol., 179: 258-265.
CrossRefDirect Link - Saber, W.I.A., M.M. El-Metwally and M.S. El-Hersh, 2010. Keratinase production and biodegradation of some keratinous wastes by Alternaria tenuissima and Aspergillus nidulans. Res. J. Microbiol., 5: 21-35.
CrossRefDirect Link - Tatineni, R., K.K. Doddapanem, R.C. Potumarthi, R.N. Vellanki, M.T. Kandathil, N. Kolli and L.N. Mangamoori, 2008. Purification and characterization of an alkaline keratinase from Streptomyces sp. Bioresour. Technol., 99: 1596-1602.
CrossRefPubMedDirect Link - Tsiroulnikov, K., H. Rezai, E.B. Osmolovskaya, P. Nedkov and A. Gousterova et al., 2004. Hydrolysis of the amyloid prion protein and nonpathogenic meat and bone meal by anaerobic thermophilic prokaryotes and Streptomyces subspecies. J. Agric. Food Chem., 52: 6353-6360.
PubMedDirect Link - Yoshioka, M., T. Miwa, H. Horii, M. Takata and T. Yokoyama et al., 2007. Characterization of a proteolytic enzyme derived from a Bacillus strain that effectively degrades prion protein. J. Applied Microbiol., 10: 509-515.
PubMedDirect Link - Bradford, M.M., 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem., 72: 248-254.
CrossRefPubMedDirect Link